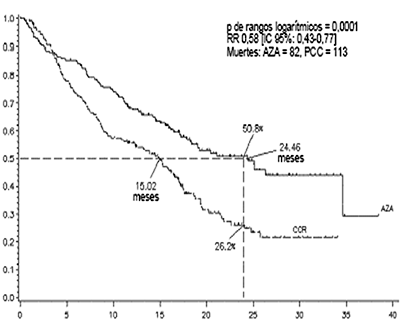VIDAZA
AZACITIDINA
Viales
1 Vial(es), 100 mg,
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA: Cada VIAL contiene:
|
Azacitidina |
100 mg |
|
Manitol |
100 mg |
Después de la reconstitución, cada mL de la suspensión contiene 25 mg de azacitidina.
FORMA FARMACÉUTICA:
Polvo para suspensión inyectable.
Polvo liofilizado blanco.
INDICACIONES TERAPÉUTICAS: VIDAZA® está indicado para el tratamiento de pacientes adultos que no se consideran aptos para el trasplante de células madre hematopoyéticas y que padecen:
Síndromes mielodisplásicos (SMD) intermedios 2 y de alto riesgo, según el sistema internacional de puntuación pronóstica (IPSS).
Leucemia mielomonocítica crónica (LMMC) con el 10 al 29% de blastos medulares sin trastorno mieloproliferativo.
Leucemia mieloide aguda (LMA) con 20 al 30% de blastos y displasia multilínea, según la clasificación de la Organización Mundial de la Salud (OMS).
DATOS FARMACÉUTICOS:
Listado de excipientes: Manitol.
Incompatibilidades: Este medicamento no debe mezclarse con otros.
PROPIEDADES FARMACOCINÉTICAS: Las propiedades farmacocinéticas de la azacitidina se estudiaron después de dosis únicas de 75 mg/m2 administradas por vías subcutánea e intravenosa.
Absorción: La azacitidina se absorbió rápidamente después de la administración por vía subcutánea; se produjeron concentraciones plasmáticas máximas de azacitidina de 750 ± 403 ng/ml a las 0,5 horas (el primer punto de extracción de muestras) después de la administración de la dosis. La biodisponibilidad absoluta de la azacitidina después de la administración por vía subcutánea en relación con la intravenosa fue de aproximadamente el 89%, basado en el área bajo la curva (AUC).
Distribución: Después de la administración por vía intravenosa, el volumen medio de distribución fue de 76 ± 26 l, y el aclaramiento sistémico fue de 147 ± 47 l/h.
Metabolismo: A partir de la información obtenida in vitro, aparentemente, el metabolismo de la azacitidina no está mediado por las isoenzimas del citocromo P450 (CYP), las UDP-glucuronosiltransferasas (UGT), sulfotransferasas (SULT) y glutatión transferasas (GST).
El metabolismo de la azacitidina es mediante hidrólisis espontánea y por desaminación mediada por la citidina deaminasa. En fracciones S9 del hígado humano, la formación de metabolitos fue independiente del NADPH, lo que implica que cualquier metabolismo sería catalizado por enzimas citosólicas. Estudios in vitro de azacitidina con hepatocitos humanos cultivados indican que, a concentraciones de 1,0 a 100 µM (es decir, hasta concentraciones aproximadamente 30 veces más altas que las que se alcanzan clínicamente), la azacitadina no induce las isoenzimas del citocromo P450 (CYP) 1A2, 2C19, 3A4 o 3A5. En un estudio para evaluar la inhibición de una serie de isoenzimas del P450 (CYP 1A2, 2C9, 2C19, 2D6, 2E1 y 3A4), incubadas con azacitidina 100 µM, no se pudieron determinar los valores de CI50; por lo tanto, es improbable la inhibición enzimática por azacitidina a concentraciones plasmáticas clínicamente alcanzables. No se ha estudiado la posibilidad de inhibir el CYP2B6 o el 2C8.
Excreción: La azacitidina se aclara rápidamente del plasma, con una semivida de eliminación (t½) media de 41 ± 8 minutos, después de la administración por vía subcutánea. La excreción urinaria es la principal ruta de eliminación de la azacitidina y/o de sus metabolitos. Después de la administración por vías intravenosa y subcutánea de 14C-Azacitidina, del 50 al 85% de la radioactividad administrada se recuperó en la orina, mientras que <1% se recuperó en las heces.
Poblaciones especiales: No se han estudiado formalmente los efectos de la insuficiencia renal o hepática, del sexo, de la edad o de la raza sobre las propiedades farmacocinéticas de la azacitidina.
Farmacogenómica: No se ha investigado formalmente el efecto de los polimorfismos conocidos de la citidina deaminasa sobre el metabolismo de la azacitidina.
CONTRAINDICACIONES:
Hipersensibilidad conocida al principio activo o a alguno de los excipientes.
Tumores hepáticos malignos avanzados.
Lactancia.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE EMPLEO:
Toxicidad hematológica: El tratamiento con azacitidina se asocia con anemia, neutropenia y trombocitopenia, especialmente en los dos primeros ciclos. Deben efectuarse recuentos sanguíneos completos cuando sea necesario para vigilar la respuesta y la toxicidad, pero por lo menos, antes de cada ciclo de tratamiento. Después de la administración de la dosis recomendada para el primer ciclo, la dosis para los ciclos posteriores debe reducirse o su administración debe retrasarse según sean el recuento nadir y la respuesta hematológica. Se debe advertir a los pacientes que comuniquen inmediatamente episodios febriles. Se aconseja a los pacientes y a los médicos que estén atentos a la presencia de signos y síntomas de hemorragia.
Insuficiencia hepática: No se han realizado estudios formales en pacientes con insuficiencia hepática. Se han notificado con rara frecuencia casos de coma hepático progresivo y muerte durante el tratamiento con azacitidina en los pacientes con una carga tumoral amplia debido a enfermedad metastásica, especialmente en los pacientes con niveles de albúmina sérica inicial < 30 g/l. La azacitidina está contraindicada en los pacientes con tumores hepáticos malignos avanzados.
Insuficiencia renal: En los pacientes tratados con azacitidina por vía intravenosa en combinación con otros fármacos quimioterapéuticos, se han notificado con rara frecuencia anomalías renales que variaron entre un aumento de la creatinina sérica e insuficiencia renal y muerte. Además, cinco pacientes con leucemia mieloide crónica (LMC), tratados con azacitidina y etopósido, desarrollaron acidosis tubular renal, definida como una disminución del bicarbonato sérico a < 20 mmol/l, asociada a orina alcalina e hipopotasemia (potasio sérico < 3 mmol/l). Si se producen disminuciones inexplicadas del bicarbonato sérico (< 20 mmol/l) o aumentos de la creatinina sérica o del NUS, la dosis debe disminuirse o la administración debe retrasarse.
Se debe vigilar atentamente la toxicidad en los pacientes con insuficiencia renal, puesto que la azacitidina y/o sus metabolitos se excretan principalmente por el riñón.
Cardiopatía y enfermedad pulmonar: Los pacientes con antecedentes de insuficiencia cardiaca congestiva grave, cardiopatía clínicamente inestable o enfermedad pulmonar fueron excluidos del ensayo clínico fundamental; por lo tanto, no se han establecido la seguridad ni la eficacia de VIDAZA® en estos pacientes.
PRECAUCIONES ESPECIALES DE CONSERVACIÓN: Este medicamento no requiere condiciones especiales de conservación.
Conservar los viales no reconstituidos a temperatura no mayor a 30 °C. Se permiten oscilaciones entre 15 y 30 °C.
Para las condiciones de conservación del medicamento reconstituido, (ver Periodo de validez).
PRECAUCIONES ESPECIALES DE ELIMINACIÓN Y OTRAS MANIPULACIONES:
Recomendaciones para una manipulación segura: VIDAZA® es un medicamento citotóxico y, al igual que con otros compuestos potencialmente tóxicos, debe tenerse precaución al manipular y preparar suspensiones de azacitidina. Deben aplicarse los procedimientos para la manipulación y eliminación correctas de medicamentos contra el cáncer. Si la azacitidina reconstituida entra en contacto con la piel, la zona deberá lavarse inmediatamente y a fondo con agua y jabón. Si entra en contacto con membranas mucosas, debe lavarse a fondo con agua.
Procedimiento de reconstitución:
1. Deben montarse los siguientes elementos:
Vial/es de azacitidina; vial/es de agua para preparaciones inyectables; guantes quirúrgicos no estériles.
Algodón; jeringas para inyección de 5 ml, con agujas.
2. Deben extraerse 4 ml de agua para preparaciones inyectables en la jeringa, asegurándose de purgar el aire atrapado dentro de la jeringa.
3. La aguja de la jeringa que contiene los 4 ml de agua para preparaciones inyectables debe introducirse a través del tapón de goma del vial de azacitidina; a continuación, se inyecta en el vial el agua para preparaciones inyectables.
4. Después de extraer la jeringa y la aguja, el vial debe agitarse vigorosamente, hasta obtener una suspensión turbia uniforme. Después de la reconstitución, cada ml de suspensión contendrá 25 mg de azacitidina (100 mg/4 ml). El producto reconstituido es una suspensión turbia y homogénea, sin aglomerados. La suspensión debe desecharse si contiene partículas grandes o aglomerados.
5. El tapón de goma debe limpiarse y se introduce una jeringa nueva con una aguja. A continuación, el vial debe invertirse, asegurándose de que la punta de la aguja esté por debajo del nivel del líquido. Seguidamente, debe tirarse del émbolo hacia atrás para extraer la cantidad de medicamento necesaria para la dosis correcta, asegurándose de purgar el aire atrapado dentro de la jeringa. A continuación, debe extraerse del vial la jeringa con la aguja y la aguja debe desecharse.
6. Seguidamente, debe ajustarse firmemente a la jeringa una aguja subcutánea nueva (se recomienda el calibre 25) para inyectables. La aguja no debe purgarse antes de la inyección, a fin de reducir la incidencia de reacciones locales en el lugar de la inyección.
7. Si es necesario (dosis superiores a 100 mg), deben repetirse todos los pasos anteriores para la preparación de la suspensión. Para las dosis superiores a 100 mg (4 ml), la dosis debe dividirse en partes iguales, en dos jeringas (p. ej., dosis de 150 mg = 6 ml; dos jeringas con 3 ml en cada jeringa).
8. El contenido de la jeringa de dosificación debe volver a resuspenderse inmediatamente antes de la administración. La temperatura de la suspensión en el momento de la inyección debe ser de aproximadamente 20 a 25 °C. Para volver a suspender, haga rodar vigorosamente la jeringa entre las palmas de las manos, hasta obtener una suspensión uniforme y turbia. La suspensión debe desecharse si contiene partículas grandes o aglomerados.
La suspensión de VIDAZA® debe prepararse inmediatamente antes de su uso y la suspensión reconstituida debe administrarse en los siguientes 45 minutos. Si el tiempo transcurrido es superior a 45 minutos, la suspensión reconstituida debe desecharse correctamente y debe prepararse una dosis nueva. O bien, si es necesario reconstituir el producto antes de la administración, debe colocarse en heladera (temperatura entre 2 y 8 °C) inmediatamente después de la reconstitución, y debe mantenerse en heladera durante ocho horas como máximo. Si el tiempo transcurrido en heladera es superior a ocho horas, la suspensión debe desecharse correctamente y debe prepararse una dosis nueva. Debe permitirse que la jeringa cargada con la suspensión reconstituida alcance una temperatura de aproximadamente 20 a 25 °C durante un tiempo máximo de 30 minutos antes de la administración. Si el tiempo transcurrido es superior a 30 minutos, la suspensión debe desecharse correctamente y debe prepararse una dosis nueva.
Cálculo de una dosis individual: La dosis total, según la superficie corporal (SC), puede calcularse de la siguiente manera:
Dosis total (mg) = dosis (mg/m2) x SC (m2)
La siguiente tabla se presenta sólo como un ejemplo para calcular dosis individuales de azacitidina, basadas en un valor promedio de SC de 1,8 m2.
|
Dosis, mg/m2 (% de la dosis inicial recomendada) |
Dosis total basada en un valor de SC de 1,8 m2 |
Número de viales necesarios |
Volumen total de suspensión reconstituida requerida |
|
75 mg/m2 (100%) |
135 mg |
2 viales |
5,4 ml |
|
37,5 mg/m2 (50%) |
67,5 mg |
1 vial |
2,7 ml |
|
25 mg/m2 (33%) |
45 mg |
1 vial |
1,8 ml |
Forma de administración: VIDAZA® reconstituido debe inyectarse por vía subcutánea (introduzca la aguja con un ángulo de 45 a 90°), con una aguja de calibre 25, en el brazo, el muslo o el abdomen.
Las dosis superiores a 4 ml deben inyectarse en dos lugares separados.
Los lugares de inyección deben someterse a rotación. Las nuevas inyecciones deben administrarse como mínimo a 2,5 cm de distancia del lugar anterior y nunca en zonas sensibles, con equimosis, enrojecidas o endurecidas.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
![]()
® Marca registrada
EMBARAZO Y LACTANCIA:
Embarazo: No existen datos suficientes sobre la utilización de azacitidina en mujeres embarazadas. Los estudios en ratones han mostrado toxicidad para la reproducción. Se desconoce el riesgo en seres humanos. A partir de los resultados de los estudios en animales y de su mecanismo de acción, la azacitidina no debe utilizarse durante el embarazo, especialmente durante el primer trimestre, a menos que sea claramente necesario. Las ventajas del tratamiento deben sopesarse frente al posible riesgo para el feto en cada caso concreto.
Las mujeres en edad fértil y los hombres deben utilizar un método anticonceptivo eficaz durante el tratamiento y hasta tres meses después del mismo.
Lactancia: Se desconoce si la azacitidina o sus metabolitos se excretan en la leche humana. Debido a las posibles reacciones adversas graves en el niño lactante, la lactancia está contraindicada durante el tratamiento con azacitidina.
Fertilidad: No hay información acerca del efecto de la azacitidina sobre la fecundidad en humanos. En los animales se han documentado reacciones adversas de la azacitidina sobre la fecundidad masculina. Se debe aconsejar a los hombres que no conciban un hijo mientras reciben tratamiento, debiendo utilizar un método anticonceptivo eficaz durante el tratamiento y hasta tres meses después del mismo. Antes de iniciar el tratamiento, debe aconsejarse a los pacientes varones que pidan asesoramiento sobre la conservación de esperma.
CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y TRASTORNOS DE LA FERTILIDAD: La azacitidina induce mutaciones de genes y aberraciones cromosómicas en sistemas bacterianos y de células de mamíferos in vitro. Se evaluó el potencial carcinogénico de la azacitidina en ratones y ratas. La azacitidina indujo tumores del sistema hematopoyético en ratones hembras, cuando se administró por vía intraperitoneal, tres veces por semana, durante 52 semanas. En ratones tratados con azacitidina administrada por vía intraperitoneal, durante 50 semanas, se observó un aumento de la incidencia de tumores en el sistema linforreticular, los pulmones, las glándulas mamarias y la piel. Un estudio de tumorogenicidad en ratas reveló un aumento de la incidencia de tumores testiculares.
En estudios de embriotoxicidad precoz en ratones revelaron una frecuencia del 44% de muerte embrionaria intrauterina (aumento de la resorción), después de una inyección única, por vía intraperitoneal, de azacitidina durante la organogénesis. Se detectaron anomalías del desarrollo del cerebro en ratones que recibieron azacitidina durante o antes del cierre del paladar duro. En ratas, la azacitidina no causó ninguna reacción adversa cuando se administró antes de la implantación; sin embargo, fue claramente embriotóxica cuando se administró durante la organogénesis. Entre las anomalías fetales causadas durante la organogénesis se encuentran las siguientes: anomalías del sistema nervioso central (exencefalia/encefalocele), anomalías de las extremidades (micromelia, pie zambo, sindactilia, oligodactilia) y otras (micrognatia, gastrosquisis, edema y anomalías de las costillas).
La administración de azacitidina a ratones machos antes del apareamiento con ratones hembras no tratadas produjo una disminución de la fecundidad y la pérdida de la progenie durante el desarrollo embrionario y posnatal posterior. El tratamiento de ratas machos produjo una disminución del peso de los testículos y de los epidídimos, disminución de los recuentos de espermatozoides, disminución de las tasas de preñez, aumento de embriones anormales y aumento de la pérdida de embriones en hembras apareadas.
REACCIONES ADVERSAS: En el 97% de los pacientes se han producido reacciones adversas consideradas posible o probablemente relacionadas con la administración de VIDAZA®.
Las reacciones adversas descritas con mayor frecuencia con el tratamiento con azacitidina fueron reacciones hematológicas (71,4%), incluyendo trombocitopenia, neutropenia y leucopenia (generalmente de grado 3 o 4); reacciones gastrointestinales (60,6%), incluyendo náuseas, vómitos (generalmente de grado 1 o 2), o reacciones en el lugar de la inyección (77,1%; generalmente de grado 1 o 2).
Las reacciones adversas graves más frecuentes (> 2%) observadas en el ensayo fundamental (AZA PH GL 2003 CL 001) y también descritas en los ensayos de apoyo (CALGB 9221 y CALGB 8921) fueron neutropenia febril (8,0%) y anemia (2,3%). Otras reacciones adversas graves notificadas con menor frecuencia (< 2%) fueron sepsis neutropénica, neumonía, trombocitopenia y reacciones hemorrágicas (p. ej., hemorragia cerebral).
La siguiente tabla contiene las reacciones adversas en las que pudo establecerse razonablemente una relación causal con el tratamiento con la azacitidina. Las frecuencias proporcionadas se basan en las observaciones durante el ensayo clínico fundamental o en dos ensayos clínicos de apoyo.
Las frecuencias se definen como: Muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); poco frecuentes (≥ 1/1.000 a < 1/100); raras (≥ 1/10.000 a < 1/1.000); muy raras (< 1/10.000), de frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Clasificación de órganos y sistemas |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Infecciones e infestaciones |
Neumonía, nasofaringitis |
Sepsis neutropénica, infección de las vías respiratorias altas, infección de las vías urinarias, sinusitis, faringitis, rinitis, herpes simple |
|
|
Trastornos de la sangre y del sistema linfático |
Neutropenia febril, neutropenia, leucopenia, trombocitopenia, anemia |
Insuficiencia medular, pancitopenia |
|
|
Trastornos del sistema inmunológico |
Reacciones de hipersensibilidad |
||
|
Trastornos del metabolismo y de la nutrición |
Anorexia |
Hipopotasemia |
|
|
Trastornos psiquiátricos |
Estado de confusión, ansiedad, insomnio |
||
|
Trastornos del sistema nervioso |
Mareos, cefalea |
Hemorragia intracraneal, letargo |
|
|
Trastornos oculares |
Hemorragia ocular, hemorragia conjuntival |
||
|
Trastornos vasculares |
Hipertensión, hipotensión, hematoma |
||
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Disnea de esfuerzo, dolor faringolaríngeo |
|
|
Trastornos gastrointestinales |
Diarrea, vómitos, estreñimiento, náuseas, dolor abdominal |
Hemorragia gastrointestinal, hemorragia hemorroidal, estomatitis, hemorragia gingival, dispepsia |
|
|
Trastornos de la piel y del tejido subcutáneo |
Petequias, prurito, exantema, equimosis |
Púrpura, alopecia, eritema, exantema macular |
|
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia |
Mialgia, dolor musculoesquelético |
|
|
Trastornos renales y urinarios |
Hematuria |
||
|
Trastornos generales y alteraciones en el lugar de administración |
Fatiga, pirexia, dolor torácico, eritema en el lugar de la inyección, dolor en el lugar de la inyección, reacción (no especificada) en el lugar de la inyección |
Lugar de la inyección: equimosis, hematoma, induración, exantema, prurito, inflamación, decoloración, nódulo y hemorragia. Malestar |
|
|
Exploraciones complementarias |
Disminución del peso |
Reacciones adversas hematológicas: Las reacciones adversas notificadas con mayor frecuencia, asociadas al tratamiento con azacitidina, fueron hematológicas, que incluyen trombocitopenia, neutropenia y leucopenia, generalmente de grado 3 o 4. Hay un mayor riesgo de que estas reacciones se produzcan en los dos primeros ciclos, después de los cuales se producen con menor frecuencia y los pacientes restablecen la función hematológica. La mayoría de las reacciones adversas hematológicas se controlaron mediante la vigilancia rutinaria de los recuentos sanguíneos completos y retrasando la administración de azacitidina en el siguiente ciclo, antibióticos profilácticos y/o apoyo con factor de crecimiento (p. ej., G-CSF) para la neutropenia, y transfusiones para la anemia o la trombocitopenia, según fuera necesario.
Infecciones: La mielosupresión puede llevar a neutropenia y a un aumento del riesgo de infección. En los pacientes que han recibido azacitidina se han notificado reacciones adversas graves, como sepsis neutropénica (0,8%) y neumonía (2,5%). Las infecciones pueden tratarse con el empleo de un antiinfeccioso y refuerzo con factor del crecimiento (p. ej., G-CSF) para la neutropenia.
Hemorragias: Puede producirse hemorragia en los pacientes que reciben azacitidina. Se han notificado reacciones adversas graves, como hemorragia digestiva (0,8%) y hemorragia intracraneal (0,5%). Se debe vigilar la presencia de signos y síntomas de hemorragia en los pacientes, sobre todo en los que presentan trombocitopenia preexistente o relacionada con el tratamiento.
Hipersensibilidad: Se han notificado reacciones de hipersensibilidad graves (0,25%) en los pacientes que recibían azacitidina. En caso de reacción anafiláctica, el tratamiento con azacitidina debe suspenderse inmediatamente y debe iniciarse el tratamiento sintomático adecuado.
Reacciones adversas de la piel y del tejido subcutáneo: La mayoría de las reacciones adversas cutáneas y del tejido subcutáneo se relacionaron con el lugar de la inyección. En el ensayo fundamental, ninguna de estas reacciones adversas llevó a la suspensión temporal o permanente del tratamiento con azacitidina, ni a la disminución de la dosis de azacitidina. La mayoría de las reacciones adversas se produjeron en los dos primeros ciclos de tratamiento y tendieron a disminuir en los ciclos posteriores. Las reacciones adversas del tejido subcutáneo, como exantema, inflamación y prurito en el lugar de la inyección, exantema, eritema y lesión cutánea pueden precisar el tratamiento con un medicamento concomitante, como antihistamínicos, corticoesteroides y medicamentos antiinflamatorios no esteroideos (AINES).
Reacciones adversas gastrointestinales: Las reacciones adversas gastrointestinales notificadas con mayor frecuencia, relacionadas con el tratamiento con azacitidina, incluyeron estreñimiento, diarrea, náuseas y vómitos. Estas reacciones adversas se trataron sintomáticamente con antieméticos para las náuseas y los vómitos, antidiarreicos para la diarrea, y laxantes y/o ablandadores de las heces para el estreñimiento.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas. Se debe advertir a los pacientes que pueden sufrir reacciones adversas, como fatiga, durante el tratamiento. Por lo tanto, debe recomendarse precaución al conducir un vehículo o utilizar máquinas.
INTERACCIÓN CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN: A partir de la información obtenida in vitro, aparentemente, el metabolismo de la azacitidina no está mediado por las isoenzimas del citocromo P450 (CYP), las UDP-glucuronosiltransferasas (UGT), sulfotransferasas (SULT) y glutatión transferasas (GST); por lo tanto, las interacciones relacionadas con estas enzimas metabolizantes in vivo se consideran improbables.
Los efectos inhibitorios o inductores clínicamente significativos de la azacitidina sobre las enzimas del citocromo P450 son improbables.
No se han realizado estudios formales de interacción farmacológica clínica con la azacitidina.
FARMACOLOGÍA CLÍNICA:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Agente antineoplásico, análogos de la pirimidina.
Código ATC: L01BC07 8.
Mecanismo de acción: Se cree que la azacitidina ejerce sus efectos antineoplásicos mediante diversos mecanismos, que incluyen citotoxicidad sobre las células hematopoyéticas anormales en la médula ósea e hipometilación del ADN. Los efectos citotóxicos de la azacitidina pueden deberse a diversos mecanismos, que incluyen la inhibición del ADN, el ARN y la síntesis de proteínas, la incorporación en el ARN y en el ADN, y la activación de las vías que causan daño en el ADN. Las células no proliferativas son relativamente insensibles a la azacitidina. La incorporación de azacitidina en el ADN produce la inhibición de las metiltransferasas de ADN, lo que lleva a la hipometilación del ADN. La hipometilación del ADN de genes metilados aberrantemente, que intervienen en las vías de regulación normal del ciclo celular, diferenciación y muerte puede producir la re-expresión de genes y el restablecimiento de funciones supresoras del cáncer en las células cancerosas. No se ha establecido la importancia relativa de la hipometilación del ADN frente a la citotoxicidad u otras actividades de la azacitidina con los desenlaces clínicos.
Eficacia y seguridad clínicas: Se estudiaron la eficacia y la seguridad de VIDAZA® en un ensayo comparativo internacional, multicéntrico, controlado, abierto, aleatorio, con grupos paralelos y de fase III (AZA PH GL 2003 CL 001), en pacientes con: SMD intermedio 2 y de alto riesgo, según el sistema internacional de puntuación pronóstica (IPSS), anemia refractaria con exceso de blastos (RAEB), anemia refractaria con exceso de blastos en transformación (RAEB-T) y leucemia mielomonocítica crónica modificada (LMMCm) según el sistema de clasificación franco-americano-británico (FAB). Actualmente, en el sistema vigente de clasificación de la OMS, se considera que los pacientes con RAEB-T (del 21 al 30% de blastos) son pacientes con LMA. Se comparó el tratamiento con azacitidina más el mejor cuidado de apoyo (BSC) (n = 179) con pautas de cuidados convencionales (PCC). Las PCC consistieron en el BSC solo (n = 105), citarabina a dosis bajas más el BSC (n = 49) o quimioterapia de inducción estándar más el BSC (n = 25). Antes de la asignación aleatoria, los pacientes fueron preseleccionados por su médico para una de las tres PCC. Los pacientes recibieron esta pauta preseleccionada si no fueron asignados aleatoriamente para recibir VIDAZA®. Como parte de los criterios de inclusión, se exigió a los pacientes que tuvieran un estado funcional de 0 a 2 del Grupo Oncológico Cooperativo del Este (Eastern Cooperative Oncology Group, ECOG). Se excluyó del ensayo a los pacientes con SMD secundarios. El criterio principal de valoración del ensayo fue la supervivencia total. VIDAZA® se administró a una dosis por vía subcutánea de 75 mg/m2 diarios, durante siete días, seguido de un periodo de reposo de 21 días (ciclo de tratamiento de 28 días), durante una mediana de nueve ciclos (intervalo = 1-39), y una media de 10,2 ciclos. Dentro de la población con intención de tratar (ITT), la mediana de edad fue de 69 años (intervalo de 38 a 88 años).
En el análisis ITT de 358 pacientes (179 que recibieron azacitidina y 179 PCC), el tratamiento con VIDAZA® se asoció a una mediana de supervivencia de 24,46 meses, en comparación con 15,02 meses en el caso de los que recibieron el tratamiento de PCC, una diferencia de 9,4 meses, con un p-valor de rangos logarítmicos estratificados de 0,0001. La razón de riesgos para el efecto del tratamiento fue 0,58 (IC 95%: 0,43-0,77). Las tasas de supervivencia a los dos años fueron del 50,8% en el caso de los pacientes tratados con azacitidina, en comparación con el 26,2% de los pacientes que recibieron PCC (p < 0,0001).
|
Proporción superviviente |
|
|
Tiempo (meses) desde la aleatorización |
|
# a riesgo
|
AZA |
179 |
152 |
130 |
85 |
52 |
30 |
10 |
1 |
|
CCR |
179 |
132 |
95 |
69 |
32 |
14 |
5 |
0 |
Leyenda:
AZA = Azacitidina;
IC = Intervalo de confianza;
PCC = Pautas de cuidados convencionales;
RR = Razón de riesgos.
Los beneficios de VIDAZA® en cuanto a la supervivencia fueron constantes, independientemente de la opción de tratamiento de PCC (BSC solo, citarabina a dosis baja más BSC o quimioterapia de inducción estándar más BSC) utilizados en el grupo control.
Cuando se analizaron los subgrupos citogenéticos de IPSS, se observaron resultados similares en cuanto a la mediana de la supervivencia total en todos los grupos (citogenético bueno, intermedio, deficiente, incluida la monosomía 7).
En los análisis de los subgrupos de edad, se observó un aumento de la mediana de supervivencia total en todos los grupos (< 65 años, ≥ 65 años y ≥ 75 años).
El tratamiento con VIDAZA® se asoció a una mediana del tiempo hasta la muerte o la transformación en LMA de 13,0 meses, en comparación con 7,6 meses en los pacientes que recibieron tratamiento de PCC, una mejoría de 5,4 meses con un p-valor de rangos logarítmicos estratificados de 0,0025.
El tratamiento con VIDAZA® también se asoció a una disminución de las citopenias y de sus síntomas relacionados. El tratamiento con VIDAZA® llevó a una disminución de la necesidad de transfusiones de hematíes y de plaquetas. De los pacientes del grupo tratado con azacitidina que eran dependientes de la transfusión de hematíes al inicio, el 45,0% de estos pacientes pasaron a ser independientes de la transfusión de hematíes durante el periodo de tratamiento, en comparación con el 11,4% de los pacientes de los grupos combinados tratados con pautas de cuidados convencionales (una diferencia estadísticamente significativa [p < 0,0001] del 33,6% [IC 95%: 22,4-44,6]). En los pacientes que dependían de transfusión de hematíes al inicio y que se convirtieron en independientes, la mediana de la duración de la independencia de la transfusión de hematíes fue de 13 meses en el grupo que recibió azacitidina.
La respuesta fue evaluada por el investigador o por un comité de revisión independiente (CRI). La respuesta total (remisión completa [RC] + remisión parcial [RP]) determinada por el investigador fue del 29% en el grupo tratado con azacitidina y del 12% en el grupo combinado con pautas de cuidados convencionales (p = 0,0001). La respuesta total (RC + RP) determinada por el CRI en el ensayo AZA PH GL 2003 CL 001 fue del 7% (12/179) en el grupo tratado con azacitidina, en comparación con el 1% (2/179) en el grupo combinado con pautas de cuidados convencionales (p = 0,0113). Las diferencias entre las evaluaciones de la respuesta por el CRI y por el investigador se produjeron a consecuencia de los criterios del Grupo Internacional de Trabajo (IWG, International Working Group), que requieren mejoría en los recuentos en sangre periférica y en el mantenimiento de estas mejorías durante un mínimo de 56 días. Se demostró también un beneficio en cuanto a la supervivencia en los pacientes que no habían logrado una respuesta completa o parcial después del tratamiento con azacitidina. Se alcanzó una mejoría hematológica (mayor o menor), como la determina el CRI, en el 49% de los pacientes que recibieron azacitidina, en comparación con el 29% de los pacientes combinados tratados con pautas de cuidados convencionales (p < 0,0001).
En los pacientes con una o más anomalías citogenéticas al inicio, el porcentaje de pacientes con una respuesta citogenética mayor fue similar al de los grupos tratados con azacitidina y con pautas combinadas de cuidados convencionales. La respuesta citogenética menor fue estadísticamente significativamente (p = 0,0015) más alta en el grupo tratado con azacitidina (34%), en comparación con el grupo combinado con pautas de cuidados convencionales (10%).
POSOLOGÍA/ADMINISTRACIÓN:
El tratamiento con VIDAZA® debe iniciarse y monitorizarse bajo la supervisión de un médico con experiencia en el uso de fármacos quimioterapéuticos. Los pacientes deben ser tratados previamente con antieméticos para las náuseas y los vómitos.
Posología: La dosis inicial recomendada para el primer ciclo de tratamiento, para todos los pacientes, independientemente de los valores hematológicos iniciales, es de 75 mg/m2 de superficie corporal, inyectada por vía subcutánea, diariamente, durante siete días, seguido de un periodo de reposo de 21 días (ciclo de tratamiento de 28 días).
Se recomienda que los pacientes reciban tratamiento durante un mínimo de seis ciclos. El tratamiento debe continuarse mientras el paciente siga beneficiándose o hasta la progresión de la enfermedad.
Se deben vigilar la respuesta/toxicidad hematológica y la toxicidad renal de los pacientes; puede ser necesario un retraso en el inicio del siguiente ciclo o una disminución de una dosis, como se explica más adelante.
Ajuste de la dosis debido a toxicidad hematológica: La toxicidad hematológica se define como el recuento sanguíneo más bajo alcanzado en un ciclo determinado (nadir), si el recuento de plaquetas disminuye a menos de 50,0 x 109/l o el recuento absoluto de neutrófilos (RAN) disminuye a menos de 1 x 109/l.
La recuperación se define como un aumento de la/s línea/s celular/es en las que se observó una toxicidad hematológica, como mínimo, igual a la mitad de la diferencia entre el nadir y el recuento inicial, más el recuento nadir; es decir, recuento sanguíneo en la recuperación ≥ recuento nadir + (0,5 x [recuento inicial - recuento nadir]).
Pacientes sin una disminución de los recuentos sanguíneos iniciales (es decir, leucocitos > 3,0 x 109/l y RAN > 1,5 x 109/l, y recuento plaquetario > 75,0 x 109/l) antes del primer tratamiento. Si se observa toxicidad hematológica después del tratamiento con VIDAZA®, el siguiente ciclo de tratamiento con VIDAZA® debe retrasarse hasta que el recuento plaquetario y el RAN se hayan recuperado. Si la recuperación se alcanza en un plazo de 14 días, no es necesario un ajuste de la dosis. Sin embargo, si la recuperación no se ha alcanzado en un plazo de 14 días, la dosis debe reducirse según la siguiente tabla. Después de las modificaciones de la dosis, la duración del ciclo debe volver a ser de 28 días.
|
Recuentos nadir |
% de la dosis en el siguiente ciclo, si la recuperación* no se alcanza en un plazo de 14 días |
|
|
RAN (x 109/l) |
Plaquetas (x 109/l) |
|
|
£ 1,0 |
£ 50,0 |
50% |
|
> 1,0 |
> 50,0 |
100% |
|
* Recuperación = recuentos ≥ recuento nadir + (0,5 x [recuento inicial - recuento nadir]). |
||
Pacientes con recuentos sanguíneos iniciales reducidos (es decir, leucocitos < 3,0 x 109/l o RAN < 1,5 x 109/l o recuento plaquetario < 75,0 x 109/l) antes del primer tratamiento. Después del tratamiento con VIDAZA®, si la disminución del recuento leucocitario del RAN o del recuento plaquetario con respecto al recuento antes del tratamiento es inferior al 50% o superior al 50%, pero con una mejoría en la diferenciación de cualquier línea celular, el siguiente ciclo no debe retrasarse y no debe efectuarse ningún ajuste de la dosis.
Si la disminución del recuento leucocitario del RAN o del recuento plaquetario es superior al 50% con respecto al recuento antes del tratamiento, y no hay mejoría en la diferenciación de líneas celulares, el siguiente ciclo de tratamiento con VIDAZA® debe retrasarse hasta que el recuento plaquetario y el RAN se hayan recuperado. Si la recuperación se alcanza en un plazo de 14 días, no es necesario un ajuste de la dosis. Sin embargo, si la recuperación no se ha alcanzado en un plazo de 14 días, debe determinarse la celularidad de la médula ósea. Si la celularidad de la médula ósea es > 50%, no debe efectuarse un ajuste de la dosis. Si la celularidad de la médula ósea es ≤ 50%, el tratamiento debe retrasarse y la dosis debe disminuirse, según la siguiente tabla:
|
Celularidad de la médula ósea |
% de la dosis en el siguiente ciclo, si la recuperación no se alcanza en un plazo de 14 días |
|
|
Recuperación* ≤ 21 días |
Recuperación* > 21 días |
|
|
15-50% |
100% |
50% |
|
< 15% |
100% |
33% |
|
* Recuperación = recuentos ≥ recuento nadir + (0,5 x [recuento inicial - recuento nadir]) |
||
Después de las modificaciones de la dosis, la duración del ciclo debe volver a ser de 28 días.
Poblaciones especiales:
Insuficiencia renal: No se han realizado estudios formales en pacientes con disminución de la función renal. Se deben vigilar atentamente las reacciones adversas en los pacientes con insuficiencia orgánica grave. Antes del tratamiento inicial, no se recomienda ninguna modificación específica con respecto a la dosis inicial en los pacientes con insuficiencia renal (p. ej., creatinina sérica inicial o nitrógeno ureico en la sangre [NUS] ≥ 2 veces superior al límite superior de la normalidad [LSN] o bicarbonato sérico inferior a 20 mmol/l); las modificaciones posteriores de la dosis deberán basarse en los valores hematológicos y de la bioquímica renal. Si se producen disminuciones inexplicadas de los niveles de bicarbonato sérico a menos de 20 mmol/l, la dosis deberá disminuirse en un 50% en el siguiente ciclo. Si se producen aumentos inexplicados de la creatinina sérica o del NUS a ≥ 2 veces superiores a los valores iniciales y superiores al LSN, el siguiente ciclo deberá retrasarse hasta que los valores vuelvan a la normalidad o a los valores iniciales, y la dosis deberá disminuirse en un 50% en el siguiente ciclo de tratamiento.
Insuficiencia hepática: No se han realizado estudios formales en pacientes con insuficiencia hepática. Se deben vigilar atentamente las reacciones adversas en los pacientes con insuficiencia orgánica hepática grave. Antes del tratamiento inicial, no se recomienda ninguna modificación específica de la dosis inicial en los pacientes con insuficiencia hepática; las modificaciones posteriores de la dosis deben basarse en los valores hematológicos. VIDAZA® está contraindicado en los pacientes con tumores hepáticos malignos avanzados.
Pacientes de edad avanzada: No se recomienda ningún ajuste específico de la dosis en los pacientes de edad avanzada. Puesto que es más probable que los pacientes de edad avanzada presenten un deterioro de la función renal, puede ser conveniente vigilar la función renal.
Niños y adolescentes: VIDAZA® no está recomendado para uso en niños menores de 18 años debido a la escasez de datos sobre la seguridad y la eficacia.
Análisis de laboratorio: Antes de iniciar el tratamiento y antes de cada ciclo de tratamiento, deben realizarse las pruebas de función hepática y determinarse la creatinina sérica. Deben efectuarse recuentos sanguíneos completos antes del inicio del tratamiento y cuando sea necesario para vigilar la respuesta y la toxicidad, pero como mínimo, antes de cada ciclo de tratamiento.
Forma de administración: VIDAZA® reconstituido debe inyectarse por vía subcutánea en el brazo, el muslo o el abdomen. Los lugares de inyección deben someterse a rotación. Las nuevas inyecciones deben administrarse como mínimo a 2,5 cm de distancia del lugar anterior y nunca en zonas sensibles, con equimosis, enrojecidas o endurecidas.
SOBREDOSIS: Se notificó un caso de sobredosis con azacitidina durante los ensayos clínicos. Un paciente sufrió diarrea, náuseas y vómitos después de recibir una dosis única por vía intravenosa de aproximadamente 290 mg/m2, casi el cuádruple de la dosis inicial recomendada.
En caso de sobredosis, se deben vigilar en el paciente los recuentos sanguíneos adecuados y debe recibir el tratamiento de apoyo que sea necesario. No existe un antídoto específico conocido para la sobredosis de azacitidina.
NATURALEZA Y CONTENIDO DEL ENVASE:
Presentación: Vial de vidrio de tipo I, incoloro, de 30 ml, sellado con tapón de cierre butil elastómero y precinto de aluminio con botón de plástico de polipropileno.
Tamaño del envase: 1 vial de 100 mg de azacitidina.
PERIODO DE VALIDEZ:
Vial de polvo sin abrir: 4 años.
Después de la reconstitución: Se ha demostrado la estabilidad química y física, en uso, del medicamento reconstituido, a 25 °C, durante 45 minutos, y a una temperatura entre 2 y 8 °C, durante ocho horas.
Desde un punto de vista microbiológico, el producto reconstituido debe utilizarse inmediatamente. Si no se utiliza inmediatamente, los tiempos y las condiciones de conservación en uso antes de su empleo son responsabilidad del usuario y no deben ser superiores a 8 horas a una temperatura entre 2 y 8 °C.