

BOTOX
TOXINA BOTULÍNICA TIPO A
Viales
, Viales,100 U
Frasco, Vial, 50 Unidades
Frasco, Vial, 200 Unidades
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICION:
BOTOX® 100 U: Cada vial contiene, ingrediente activo: Clostridium botulinum Tipo A 100 U complejo de neurotoxina (900kD), equivalente en peso a 4.8 ng de neurotoxina. Excipientes 0.5 mg de Albúmina Humana y 0.9 mg de Cloruro de Sodio.
BOTOX® BTX-A® 50U: Cada vial contiene, ingrediente activo: Clostridium botulinum Tipo A complejo de neurotoxina 50 U (900kD). Excipientes: 0.25 mg de Albúmina Humana y 0.45 mg de Cloruro de Sodio.
BOTOX® 200U: Cada vial contiene, ingrediente activo: Clostridium botulinum Tipo A complejo de neurotoxina 200 U (900kD). Excipientes 1.0 mg de Albúmina Humana y 1.8 mg de Cloruro de Sodio.
FORMA FARMACEUTICA
Polvo estéril secado al vacio para reconstituir a solución inyectable.
INDICACIONES
|
BOTOX® BTX-A® 50U |
BOTOX® 200U |
BOTOX® 100U |
|
Tratamiento de la hiperactividad muscular por su acción como agente inhibidor de liberación de acetilcolina presináptica, en las patologías: Oftalmología: • Blefaroespasmo esencial benigno o asociado a distonía • Estrabismo • Distonía focal Neurología: • Parálisis cerebral • Tremor • Espasticidad • Distonías • Mioclonias • Espasmo hemifacial • Cefalea tensional • Tortícolis Espasmódica. Urología: • Hiperactividad del músculo detrusor de la vejiga. Otorrinolaringología: • Temblor palatal esencial • Disfonía espasmódica. Dermatología: • Hiperhidrosis refractaria a Tratamientos convencionales. • Tratamiento de Líneas Faciales Hiperfuncionales Traumatología/Ortopedia: • Padecimientos espásticos, dolor en espalda, cuello y espina dorsal asociados a contracturas patológicas. Alternativo en la Profilaxis del Dolor de Cabeza en Migraña Crónica |
Tratamiento de la hiperactividad muscular en las patologías abajo relacionadas, por su acción como agente inhibidor de la liberación de acetilcolina presináptica: Oftalmología: • Blefaroespasmo esencial benigno o asociado a distonía • Estrabismo • Distonía focal Neurología: • Parálisis cerebral • Tremor • Espasticidad • Distonías • Mioclonias • Espasmo hemifacial • Cefalea tensional • Tortícolis Espasmódica. Urología: • Hiperactividad del músculo detrusor de la vejiga. Otorrinolaringología: • Temblor palatal esencial • Disfonía espasmódica. Dermatología: • Hiperhidrosis Focal Axilar y Palmar • Tratamiento de Líneas Faciales Hiperfuncionales Traumatología/Ortopedia: • Padecimientos espásticos, dolor en espalda, cuello y espina dorsal asociados a contracturas patológicas. Alternativo en la Profilaxis del Dolor de Cabeza en Migraña Crónica |
Tratamiento de la hiperactividad muscular en las siguientes patologías: Para Botox 100 no esta Oftalmología: - Blefaroespasmo esencial benigno o asociado a distonia - Estrabismo - Distonia focal. Neurología: - Coadyuvante o alternativo en parálisis cerebral - Tremor esencial que no ha respondido a otros tratamientos orales - Espasticidad - Distonias - Mioclonías que cursen con fenómenos distónicos - Espasmo hemifacial; - Cefalea tensional; - Tortícolis espasmódica. Urología: - Hiperactividad del músculo detrusor de la vejiga. Otorrinolaringología: - Temblor palatal esencial; - Disfonía espasmódica. Dermatología: - Hiperhidrosis refractaria a tratamientos convencionales. Traumatología/Ortopedia: - Coadyuvante en padecimientos espásticos; - Dolor de cuello y espina dorsal asociado a contracturas patológicas que no han respondido a ninguna otra medida terapéutica - Bruxismo temporo-maxilar. Proctología: Fisura anal. Gastroenterología: Acalasia en casos de que no pueda hacerse dilatación neumática o cirugía; Tratamiento de líneas faciale hiperfuncionales Alternativo en la Profilaxis del Dolor de Cabeza en Migraña Crónica |
USO GERIÁTRICO: En general, los estudios clínicos de BOTOX® no identifican diferencias en la repuesta entre los pacientes ancianos y los más jóvenes. La selección de la dosis para un paciente de edad avanzada deberá ser cuidadosa, comenzando usualmente en el extremo inferior del rango de dosis.
Para información específica sobre la indicación, consultar la Sección 5, cuando aplique.
PROPIEDADES FARMACOCINETICAS
Características generales del ingrediente activo:
Estudios de distribución en ratas indican una difusión muscular mínima del complejo de neurotoxina botulínica tipo A marcado con 125I en el músculo gastrocnemio, después de la inyección, seguida de un metabolismo sistémico rápido y de la excreción urinaria. La cantidad de material marcado radiactivamente en el músculo disminuyó con tiempo de vida media aproximado de 10 horas. En el sitio de inyección, el material radiactivo estuvo sobre todo en forma de macromoléculas de gran tamaño, mientras que muy poca de la radiactividad que alcanzó la circulación sistémica fue precipitable con TCA, lo cual sugiere una exposición sistémica mínima a la toxina después de la inyección del complejo de neurotoxina botulínica tipo A marcado con 125I en el músculo gastrocnemio. Dentro de las 24 horas posteriores a la administración, 60% de la radiactividad fue excretada en la orina. Es probable que la toxina sea metabolizada por proteasas y que los componentes moleculares sean incorporados a rutas metabólicas normales.
No se han realizado estudios clásicos de absorción, distribución, biotransformación y eliminación del ingrediente activo, debido a la naturaleza de este producto.
Características en los pacientes:
Se piensa que la distribución sistémica de las dosis terapéuticas de BOTOX® es sumamente limitada. No se espera que BOTOX® esté presente en la sangre periférica en niveles medibles después de la inyección IM o intradérmica en las dosis recomendadas. No se espera que las cantidades recomendadas de neurotoxina, administradas en cada sesión de tratamiento traigan consigo manifestación de efectos clínicos distantes de tipo sistémico (debilidad muscular) en pacientes que no presentan otra disfunción neuromuscular. Sin embargo, estudios clínicos que han utilizado técnicas electromiográficas de fibra única, han mostrado hallazgos electrofisiológicos sutiles concordantes con inhibición neuromuscular (inestabilidad o “jitter”) en músculos lejanos al sitio de inyección, pero los mismos no han estado acompañados de signos o síntomas clínicos de inhibición neuromuscular causada por los efectos de la toxina botulínica.
PROPIEDADES FARMACOLOGICAS
El Complejo de Neurotoxina BOTOX® (toxina botulínica tipo A) se produce por la fermentación de Clostridium botulinum tipo A (Cepas Hall) y se purifica a partir de la solución de cultivo en un complejo de peso molecular aproximado de 900kD que consiste de la neurotoxina y varias proteínas accesorias. El complejo se disuelve en solución de cloruro de sodio estéril que contiene albúmina sérica humana y se filtra de forma estéril (0,2 micras) antes del llenado y del secado al vacío.
El procedimiento de liberación primario para BOTOX® es un ensayo de potencia basado en células, para determinar la potencia relativa a un estándar de referencia. La prueba es específica del
producto de Allergan, BOTOX®. Una unidad de BOTOX® corresponde a la dosis letal intraperitoneal media (LD 50) calculada para ratones, en una prueba de potencia con ratones. Debido a los detalles específicos del método tales como el vehículo, el esquema de dilución y los protocolos de laboratorio, las unidades de actividad biológica de BOTOX® no pueden compararse o convertirse a unidades de cualquier otra actividad de toxina botulínica. La actividad específica de BOTOX® es aproximadamente 20 unidades/nanogramo de complejo proteico de neurotoxina.
PROPIEDADES FARMACODINAMICAS
Mecanismo de Acción: El complejo de neurotoxina de Clostridium Botulinum tipo A bloquea la liberación periférica de acetilcolina en los terminales presinápticos del nervio colinérgico, escindiendo SNAP-25, una proteína fundamental para el acoplamiento y la liberación exitosas de acetilcolina de las vesículas situadas en las terminales nerviosas.
Después de la inyección, se produce inicialmente una unión rápida de alta afinidad de la toxina a receptores específicos de la superficie de la célula. Esto es seguido por la transferencia de la toxina, a lo largo de la membrana plasmática por endocitosis, mediada por receptor. Finalmente la toxina de cadena liviana es liberada hacia el citosol donde ésta escinde SNAP-25. Este último proceso viene acompañado de la inhibición progresiva de la liberación de acetilcolina; los signos clínicos usualmente
se manifiestan en 2-3 días, con efecto máximo a las 5-6 semanas después de la inyección.
La recuperación después de la inyección intramuscular tiene lugar normalmente dentro de las 12 semanas siguientes a la inyección, a medida que nuevos terminales nerviosos brotan y permiten la
reconexión de la neurona con las placas terminales.
BOTOX® bloquea la liberación de neurotransmisores relacionados con el origen del dolor. El mecanismo que se presume para la profilaxis de la cefalea consiste en bloquear las señales periféricas
hacia el sistema nervioso central, lo cual inhibe la sensibilización central, tal como lo confirman estudios pre-clínicos y clínicos .
Después de la inyección intradetrusor, BOTOX® afecta las vías eferentes de actividad del detrusor a través de la inhibición de la liberación de acetilcolina. En adición, BOTOX® inhibe a los neurotransmisores y vías sensoriales aferentes.
Blefaroespasmo/espasmo hemifacial: Un estudio clínico aleatorizado, multicéntrico, doble ciego y de grupos paralelos que comparó la seguridad y la eficacia de 2 formulaciones de BOTOX® (con ingrediente activo derivado de diferentes Bancos de Células Maestros, una de las cuales es la actual formulación de BOTOX®) fue conducido en pacientes con blefaroespasmo esencial benigno. La dosis máxima permitida fue 100 U. Se enroló a 98 pacientes en el estudio (48 en el grupo que recibió la formulación actual de BOTOX®). En forma comparable con la formulación anterior de BOTOX®, la tasa de éxito del tratamiento con la formulación actual de BOTOX® fue de aproximadamente 90% en la semana 4.
En un estudio abierto, a 56 pacientes con espasmo hemifacial, se les inyectó una dosis inicial de 10 a 50 U, la cual fue seguida por un periodo de 22 semanas de observación. Todos los 56 pacientes presentaron mejoría y 62,5% (35/56) presentaron una mejoría notoria.
Estrabismo: En una amplia revisión de casos retrospectivos, se evaluaron 677 pacientes adultos con estrabismo tratados con una o más inyecciones de BOTOX®. 55% de estos pacientes mejoraron a una alineación de 10 dioptrías de prisma o menos cuando se evaluaron a 6 meses o más después de la inyección.
Distonia Cervical: Se condujo un estudio aleatorizado, doble ciego, controlado con placebo, multicéntrico, en pacientes con distonía cervical (CD). Dicho estudio enroló a pacientes adultos con CD y con un historial de respuesta positiva al tratamiento con BOTOX®. En total, se administró un máximo de 360 U de
BOTOX® a los pacientes.
Se evaluó a 214 pacientes en el periodo abierto, 170 de ellos fueron aleatorizados para pasar a la porción con doble ciego del estudio. Las variables de eficacia primarias fueron el cambio de la posición de la cabeza (medido a través de la Escala de Severidad de la Distonía Cervical) y el porcentaje de pacientes que mostraron cualquier mejoría del estatus de su CD (medida a través de la Escala de Evaluación Global del Médico) 6 semanas después del tratamiento. El dolor, el cual es un síntoma importante de la CD, fue evaluado por los pacientes en términos de su frecuencia y su intensidad. BOTOX® fue significativamente superior clínica y estadísticamente al placebo en términos de ambas variables primarias de la eficacia al cabo de 6 semanas. BOTOX® también fue mejor de manera significativa estadísticamente que el placebo en términos de la reducción de la intensidad y la frecuencia del dolor.
Un estudio adicional de diseño doble ciego, aleatorizado y cruzado evaluó la seguridad y la eficacia de 2 formulaciones de BOTOX® (con sustancia activa derivada de diferentes Bancos de Células Maestros, una de las cuales es la formulación actual de BOTOX®); el estudio se realizó en pacientes con CD. Se incluyó a 135 pacientes en este estudio. Los resultados corroboraron que se observó una mejoría clínica máxima en la semana 6 después de la inyección de BOTOX®. La disminución media del puntaje
total en la escala TWSTRS representó una mejoría de 35% con respecto a los valores basales y la disminución media del puntaje de discapacidad y dolor representó una disminución de 50% con
respecto a la línea basal en este punto en el tiempo. La evaluación global por parte de médicos y pacientes también demostró el efecto positivo del tratamiento con la formulación actual de BOTOX®, ya
que más del 85% de los médicos y 80% de los pacientes reportaron éxito del tratamiento en la semana 6.
Parálisis Cerebral Pediátrica:
Espasticidad de las extremidades superiores: En dos estudios aleatorizados, ciegos para el evaluador, se comparó la administración de BOTOX® junto a la atención médica estándar por sí sola en 72 niños con parálisis cerebral hemipléjica y espasticidad de las extremidades superiores. Los músculos del brazo y de la mano que fueron inyectados en este estudio incluyeron el bíceps, braquial, braquiorradial, flexor ulnar del carpo, flexor radial del carpo, pronador redondo, pronador cuadrado, flexor profundo de los dedos, flexor superficial de los dedos, flexor largo del pulgar, flexor corto del pulgar, aductor del pulgar y los interóseos.
En el estudio a 6 meses (n = 42; edad, 2 a 8 años), la espasticidad medida a través del Puntaje de Ashworth Modificado (MAS) se redujo significativamente en los niños tratados con BOTOX® (1-2 U/kg/músculo, dosis máxima = 240 U, dosis media = 137 U) en los meses 1 y 3 pero regresó a los valores basales en el mes 6. Los niños tratados con BOTOX® presentaron una mejoría significativamente superior de la función, medida a través de la Escala de Consecución de Metas (GAS). Los niños tratados con BOTOX® mejoraron más rápidamente que los niños que recibieron atención estándar por sí sola. No se reportaron efectos adversos relacionados con el tratamiento.
En el estudio de tres meses (n = 30, edad: 5-15 años), la función de las extremidades superiores fue medida utilizando la Evaluación de la Función Unilateral de las Extremidades Superiores de Melbourne. Los niños del grupo de tratamiento con BOTOX® (0.5 a 2.0 U/kg/músculo, dosis máxima = 250, dosis media = 153 U) presentaron una mejoría de la función de 14% en comparación con la ausencia de cambio en los niños del grupo de control en el mes 3 (P = 0.002). En este estudio no hubo diferencia entre grupos de tratamiento en la escala GAS.
Espasticidad de las extremidades inferiores – Pie equino: Se condujo un estudio de 3 meses de duración de diseño doble ciego, controlado con placebo y de grupos paralelos en niños con parálisis cerebral de 2 a 16 años de edad con tobillo en posición equina. 72 niños recibieron 4 U/kg de peso corporal de BOTOX® en las cabezas medial y lateral del gastrocnemio en la línea basal (2 U/kg/músculo) en el caso de los pacientes hemipléjicos y 1 U/kg/músculo en el caso de los pacientes dipléjicos, y nuevamente al cabo de 4 semanas. La dosis acumulativa de BOTOX® a lo largo de 4 semanas fue de 2-4 U/kg/músculo (8 U/kg de peso corporal en total; máximo: 200 U por visita) durante un periodo de 30 días. BOTOX® fue significativamente más eficaz que el placebo con base en una mejoría de = 2 U en el puntaje compuesto de marcha dinámica de la Escala de Calificación del Médico (PRS) (patrón de marcha, posición del tobillo, posición del pie delantero al impactar el pie, posición de la rodilla durante la marcha, grado de acuclillamiento y velocidad de marcha).
En el seguimiento abierto a largo plazo de 39 meses de duración de estos pacientes, los músculos gastrocnemios mediales y laterales recibieron la inyección de 2 U/kg/músculo (dosis total máxima: 200 U de BOTOX®) en las cabezas medial y lateral del gastrocnemio, repitiéndose la administración en lo sucesivo cada que ello fuese necesario. De 207 pacientes evaluados, 155 recibieron seguimiento durante 12 meses, 100 por 18 meses, 42 por 2 años y 7 por un máximo de 3 años. El porcentaje de pacientes que mostraron una mejoría con base en el patrón de marcha dinámica (escala PRS) varió entre 41% y 67% a lo largo del periodo de 3 años. En lo que respecta a las evaluaciones individuales incluidas en la escala PRS, se observaron mejorías significativas en cada visita a lo largo del periodo de 3 años.
Espasticidad de las extremidades inferiores – aductores de la cadera: Estudios publicados mostraron que BOTOX® es eficaz para reducir el dolor y la espasticidad y para mejorar la función. Un estudio doble ciego controlado con placebo (n = 16) en niños de 2 a 10 años de edad, encontró que la administración de 4 U de BOTOX®/kg de peso corporal a cada grupo de músculos aductores (dosis total: 8 U/kg de peso corporal) 5 -10 días antes de una cirugía programada aislada de los aductores redujo significativamente los puntajes de dolor (reducción de 74%, p = 0.003), la necesidad de analgésicos (reducción de 50%, p = 0.005) y la duración de la estadía hospitalaria (reducción de 33%, p = 0.003) en comparación con el placebo.
Un segundo estudio (n = 43, edad media: 8.2 ± 2.5 años) comparó la eficacia de BOTOX® (300 U inyectadas en los músculos aductores y en los isquiotibiales mediales) vs. una férula de presión en términos de la espasticidad de los aductores de la cadera. Ambos grupos mejoraron durante el periodo del estudio; in embargo, en la visita realizada tras 3 meses, BOTOX® fue significativamente más eficaz que las férulas de presión en términos de la espasticidad y del desempeño motor medido a través del Puntaje de Ashworth Modificado (p = 0.002) y de la distancia de la rodilla (p = 0.02).
Espasticidad focal de las extremidades superiores asociada con accidente cerebrovascular en adultos: Allergan ha completado 13 estudios sobre espasticidad de las extremidades superiores posterior a accidente cerebrovascular, incluyendo 8 estudios doble ciego controlados con placebo y 5 estudios de extensión/abiertos. En dichos estudios se enroló a 900 pacientes con espasticidad de las extremidades superiores posterior a accidente cerebrovascular; 840 pacientes que conformaron la población “todos los tratados con BOTOX®” recibieron dosis de 75 U a 400 U y 211 pacientes recibieron placebo. Los 13 estudios evaluaron el tono del flexor de la muñeca, 12 estudios evaluaron el
tono de los flexores de los dedos y 5 estudios demostraron mejoría estadística y clínicamente significativa del tono de los flexores medido a través de la Escala de Ashworth (una medida del efecto
fisiológico sobre los músculos) y de la Evaluación Global del Médico (una medida del efecto clínico del tratamiento sobre el estatus general del paciente). Tanto análisis integrados de los estudios controlados como los resultados por estudio, indican que BOTOX® es un tratamiento seguro, bien tolerado y eficaz de la espasticidad muscular focal.
Un análisis combinado de los estudios doble ciego controlados con placebo mostró una mayor mejoría de los puntajes de Ashworth del codo y de los dedos en los pacientes tratados con BOTOX® en
comparación con los que recibieron un placebo. Los resultados fueron similares en ambos grupos de edad, para ambos sexos y en ambos grupos de raza.
Espasticidad focal de las extremidades inferiores (tobillo) asociada con accidente cerebrovascular en adultos: Se realizó un estudio de Fase 3, doble ciego, aleatorizado y controlado con placebo en pacientes adultos que hubieran sufrido un accidente cerebrovascular y presentaran espasticidad de las extremidades inferiores con afectación del tobillo. Se aleatorizó un total de 120 pacientes para recibir BOTOX® (n=58) (dosis total de 300 unidades) o placebo (n=62).
Se observó una mejoría significativa en comparación con placebo para la variable principal de valoración de cambio general con respecto a la línea base hasta la semana 12 en el puntaje de la escala modificada de Ashworth para el tobillo (MAS), calculada usando el método de área bajo la curva. También se observó una mejoría significativa en comparación con el placebo para el cambio medio con respecto a la línea base para el tobillo, según el puntaje de MAS, en consultas individuales después el tratamiento en las semanas 4, 6 y 8. La proporción de pacientes que respondieron al tratamiento (con por lo menos un grado de mejoría) también fue mayor que en el grupo de pacientes tratados con placebo. También se asoció el tratamiento con BOTOX® se asoció con una mejoría significativa en la impresión clínica global del investigador (CGI) de las discapacidad funcional en
comparación con el grupo placebo. La reducción del tono muscular fue significativa, como lo demostró la correlación visible entre las evaluaciones globales de los investigadores y los pacientes.
Disfonía espasmódica: En la serie a mayor escala reportada, 639 pacientes con disfonía espasmódica aductora y 108 pacientes con disfonía espasmódica abductora recibieron la inyección de una dosis media de 3.1 ± 3.1 U y 2.16 ± 1.07 U de BOTOX®, respectivamente. Los pacientes registraron las respuestas en un diario,
incluyendo su porcentaje de función normal en una escala análoga visual global en la cual 100% representó una voz normal y 0% representó incapacidad para vocalizar. La duración media del beneficio fue de 10.5 ± 12.2 semanas y el porcentaje de función normal se elevó de 54.8% ± 21.9% a 66.7% ± 23.4%.
En otra serie a gran escala compuesta por 169 pacientes (disfonía espasmódica aductora en 88.8%, disfonía espasmódica abductora en 1.8% y disfonía espasmódica mixta en 4.1%), el puntaje mediano de resultado del tratamiento fue excelente en 63.9% de los pacientes, muy bueno en 18%, satisfactorio en 14.5% e insatisfactorio en 3.5%, con base en una escala subjetiva de autocalificación registrada por los pacientes en un diario. Una nasoendoscopía —realizada en numerosos pacientes antes y después del tratamiento— confirmó la relajación de las cuerdas vocales y la eliminación de los espasmos.
Hiperhidrosis:
Hiperhidrosis primaria de las axilas: Se condujo un estudio clínico doble ciego, multicéntrico y de un solo ciclo de tratamiento en pacientes con hiperhidrosis axilar primaria bilateral persistente definida como una medición gravimétrica basal de producción espontánea de sudor de al menos 50 mg en cada axila a lo largo de 5 minutos a temperatura ambiente en el reposo. 320 pacientes fueron aleatorizados para recibir 50 U de BOTOX® (n = 242) o placebo (n = 78). Los sujetos con respuesta al tratamiento fueron definidos como aquellos que mostraron una reducción de la sudoración axilar de al menos 50% con respecto a la línea basal. En el punto en el tiempo primario, la semana 4 posterior a la inyección, la tasa de respuesta en el grupo de tratamiento con BOTOX® fue de 93.8% en comparación con 35.9% en el que recibió el placebo (p <0.001).
Un estudio abierto de seguimiento enroló a 207 pacientes elegibles que recibieron hasta 3 tratamientos con BOTOX®. En total, 174 pacientes completaron los 16 meses de duración de los 2 estudios combinados. Se observó una tasa de respuesta elevada después de cada tratamiento con BOTOX®, con una incidencia general, en la semana 4 posterior a la inyección de sujetos con respuesta, de 91.8% después del primer tratamiento y de 88.2% después del segundo.
Se condujo otro estudio doble ciego controlado con placebo en pacientes con hiperhidrosis axilar primaria persistente. El porcentaje de sujetos con respuesta en ambos grupos de tratamiento con BOTOX® , 50 U fue 54.8% y con 75 U, 49.1%), frente al grupo que recibió placebo, 5.6% (P <0.001), pero no fue significativamente diferente al comparar ambas dosis de BOTOX®.
En un estudio abierto en una población de adolescentes de 12 a 17 años de edad (n = 144), los resultados fueron comparables a los observados en la población adulta estudiada con anterioridad.
Hiperactividad del músculo detrusor de la vejiga
Vejiga Hiperactiva: Se realizaron dos estudios clínicos multicentro, aleatorizados, doble ciego, de 24 semanas de duración, controlados con placebo, fase 3 en pacientes con vejiga hiperactiva (OAB) que presentaban síntomas de incontinencia, urgencia y frecuencia urinaria. Se distribuyeron aleatoriamente un total de 1105 pacientes cuyos síntomas no habían sido manejados adecuadamente con terapia anticolinérgica (respuesta inadecuada o efectos secundarios intolerables), para recibir o bien 100 Unidades de BOTOX (n=557) o placebo (n=548).
En ambos estudios se observaron mejorías para BOTOX 100 Unidades en comparación con placebo en la primera variable de eficacia con respecto a la línea de base, a saber, la frecuencia
diaria de episodios de incontinencia urinaria en el primer intervalo a la semana 12, incluyendo la proporción de pacientes secos. Usando la escala de beneficios del tratamiento, la proporción de pacientes que reportó una respuesta positiva al tratamiento fue significativamente mayor en el grupo de BOTOX® comparado con el grupo placebo en ambos estudios. Las mejorías significativas en
comparación con el placebo fueron también observadas para la frecuencia diaria de micción, urgencia y episodios de nocturia. El volumen evacuado por micción fue también significativamente más alto. Se observaron mejorías en todos los síntomas de OAB desde la semana 2. El tratamiento con BOTOX® en comparación con el placebo fue asociado con mejora significativa en la calidad de
vida relacionada con la salud.
Se evaluaron un total de 834 pacientes en un estudio de extensión a largo plazo. Para todas las variables de eficacia, los pacientes experimentaron una respuesta consistente con los nuevos tratamientos. La duración media de la respuesta después del tratamiento con BOTOX®, basados en la solicitud del paciente por un nuevo tratamiento fue 166 días (~24 semanas). La calificación para el re-tratamiento requirió al menos 2 episodios de incontinencia urinaria en 3 días. Ninguno de los 615 (0%) acientes con muestras analizadas desarrollaron la presencia de anticuerpos neutralizantes en suero para BOTOX®.
De 1242 pacientes tratados con BOTOX® paraVejiga Hiperactiva en estudios clínicos controlados por placebo, 41.4% (n=514) fueron de 65 años de edad o mayores, y 14.7% (n=182) fueron de 75 años de edad o mayores. No se observaron diferencias globales en el perfil de seguridad después del tratamiento con BOTOX® entre pacientes de 65 años y mayores en comparación con pacientes más jóvenes en estos estudios, con excepción de la infección del tracto urinario donde la incidencia fue más alta en pacientes de 65 años de edad o mayores tanto en el grupo de BOTOX® como en el placebo en comparación con los pacientes más jóvenes. De manera similar, no se observaron diferencias globales en efectividad entre estos grupos de edad en estudios clínicos pivotales controlados por placebo.
Hiperactividad Neurogénica del Detrusor: Se condujeron dos estudios clínicos doble ciego, controlados con placebo, aleatorizados y multicéntricos de Fase 3 en pacientes con incontinencia urinaria debida a hiperactividad neurogénica del detrusor que presentaban micción espontánea o estaban cateterizados.
Se enroló a un total de 691 pacientes con lesión de la médula espinal o esclerosis múltiple en los que no se lograba un manejo adecuado usando al menos un agente anticolinérgico. Los pacientes fueron aleatorizados para recibir 200 U de BOTOX® (n = 227), 300 U de BOTOX® (n = 223) o un placebo (n = 241).
En ambos estudios de Fase 3, se observaron mejorías significativas en comparación con el placebo en términos de la variable de eficacia primaria, cambio de la frecuencia semanal de episodios de incontinencia con respecto a la línea basal, con el tratamiento con BOTOX® (200 U y 300 U) en el punto en el tiempo primario para evaluación de la eficacia (semana 6), incluyendo el porcentaje de pacientes “secos”. Se observaron mejorías significativas de los parámetros urodinámicos incluyendo un incremento de la capacidad cistométrica máxima y disminuciones de la presión pico del detrusor durante la primera contracción involuntaria del detrusor. Asimismo, se observaron mejorías significativas de los puntajes de calidad de vida relacionada con la salud específicos para incontinencia reportados por el paciente, medidos a través del cuestionario de Calidad de Vida para Incontinencia (IQOL) (el cual incluye la conducta de evitación/limitante, el impacto psicosocial y la vergüenza social).
No fueron demostrados beneficios adicionales con BOTOX ® 300 U en comparación con 200 U de BOTOX®.
La duración media de respuesta en los dos estudios fundamentales, basados sobre la solicitud del paciente de un nuevo tratamiento, fue 256-295 días (36-42 semanas) para el grupo de 200 U comparado con 92 días (13 semanas) con placebo.
Para todos los puntos finales de eficacia, los pacientes experimentaron una respuesta consistente con el nuevo tratamiento.
En los estudios pivotales (300 Unidades y 200 Unidades), ninguno de los 475 pacientes con hiperactividad neurogénica del detrusor, con muestras analizadas, desarrollaron la presencia de anticuerpos neutralizantes. En los pacientes que continuaron bajo estudio durante un total de 4 años, incluido el estudio de extensión de etiqueta abierta, de los 383 pacientes a los que se les analizaron especímenes, 8 (2,1%) de ellos desarrollaron anticuerpos neutralizantes después de los tratamientos de repetición. 4 de Estos pacientes siguieron presentando beneficios clínicos.
Alternativo en la Profilaxis del Dolor de Cabeza en Migraña Crónica: BOTOX® fue evaluado en dos estudios multinacionales y multicéntricos de 56 semanas que incluyeron una fase doble ciego de 24 semanas de 2 ciclos de inyección que comparó a BOTOX® vs. un placebo y que fue seguida de una fase abierta de 32 semanas de 3 ciclos de inyección. En total, 1,384 adultos con migraña crónica que jamás habían recibido profilaxis para cefalea o que no utilizaron profilaxis concurrente contra cefalea durante un periodo basal de 28 días, con >15 días de cefaleas (50% de ellas migrañas o probables migrañas) y con >4 episodios de cefalea fueron estudiados en 2 estudios clínicos de Fase 3. Estos pacientes fueron aleatorizados para recibir un placebo o 155 unidades – 195 unidades de BOTOX® por inyección cada 12 semanas en la fase doble ciego de 2 ciclos. Los pacientes pudieron recibir 3 ciclos adicionales en forma abierta para un máximo de 5 ciclos de inyección. Los pacientes tuvieron permitido utilizar tratamientos agudos para cefalea (65.5% utilizó tratamientos agudos en exceso durante el periodo basal). El tratamiento con BOTOX® produjo mejorías estadísticamente (p <0.001) y clínicamente significativas respecto a la línea basal en comparación con el placebo en términos de una reducción de 50% de los días de cefalea, de la frecuencia media de días de cefalea
moderada/severa y del número total acumulativo de horas de cefalea en los días de cefalea. Los resultados de los cuestionarios HIT-6 (Prueba de Impacto de la Cefalea) y MSQ (Calidad de Vida Específica para Migraña) indicaron que BOTOX® tuvo una duración de acción sostenida y mejoró el funcionamiento, la vitalidad, la angustia psicológica y la calidad de vida general.
Líneas Faciales Hiperfuncionales:
Líneas Glabelares: En dos estudios multicéntricos, doble ciego, controlados con placebo y de grupos paralelos de diseño idéntico, pacientes con líneas glabelares moderadas a severas evaluadas en el fruncimiento máximo fueron aleatorizados para recibir BOTOX® (n = 405) o un placebo (n = 132). En estos estudios, la severidad de las líneas glabelares se redujo significativamente hasta por 120 días en el grupo de tratamiento con BOTOX® en comparación con el grupo que recibió el placebo con base en la calificación de la severidad de las líneas glabelares en fruncimiento máximo y en el reposo por parte del investigador, así como en la evaluación global del cambio de la apariencia de las líneas glabelares por parte del sujeto.
Un tercer estudio abierto fue conducido para apoyar la eficacia continua de inyecciones repetidas de BOTOX®. Una vez completados los estudios doble ciego, los pacientes tuvieron la opción de ingresar a este estudio abierto en el cual se administraron tratamientos repetidos a intervalos de 120 días. El efecto terapéutico se mantuvo a lo largo de los tres ciclos de inyección evaluados, mostrando en los resultados, un aumento de la eficacia después de múltiples sesiones de inyección.
Líneas en la frente: La inyección de BOTOX® redujo la severidad de las líneas horizontales en la frente hasta por 24 semanas. Dos semanas después de la inyección, aproximadamente 90% de los pacientes tratados con BOTOX® fueron considerados por los investigadores como sujetos con respuesta al tratamiento, y 75-80% de los pacientes consideró haber presentado mejoría. Mayores dosis de BOTOX® produjeron mayor eficacia y mayor duración del efecto.
Líneas del canto lateral (patas de gallo): La inyección de BOTOX® en el área orbitaria lateral, redujo la severidad de las arrugas en esta área (“patas de gallo”) hasta por 16 semanas. Cuatro semanas después de la inyección, 89-95% de los pacientes del grupo de tratamiento con BOTOX® fueron considerados por los investigadores como sujetos con respuesta al tratamiento, y 60-80% de los pacientes consideró que el tratamiento fue exitoso (grupos de tratamiento con 6-18 U). No hubo una diferencia significativa entre dosis. Los beneficios de una segunda inyección duraron más que los de la primera.
USO PEDIATRICO: Ha habido reportes espontáneos raros de muerte, en ocasiones asociada con neumonía por aspiración, en niños con parálisis cerebral severa después del tratamiento con toxina botulínica. No se ha establecido una asociación causal con BOTOX® en estos casos. Ha habido reportes posteriores a la comercialización sumamente raros de posibles efectos distantes del sitio de inyección en pacientes pediátricos con comorbilidades, predominantemente con parálisis cerebral, que han recibido >8 U/kg. Se deberá tener precaución extrema al tratar a pacientes pediátricos con debilidad neurológica significativa, disfagia o historial reciente de neumonía por aspiración o enfermedad pulmonar.
La seguridad y la eficacia de BOTOX® no han sido establecidas en niños menores de 2 años para la indicación de parálisis cerebral, en niños menores de 12 años para las indicaciones de blefaroespasmo, espasmo hemifacial, estrabismo, disfonía espasmódica o hiperhidrosis, en pacientes menores de 16 años para la indicación de distonía cervical ni en pacientes menores de 18 años para las indicaciones de espasticidad de las extremidades superiores e inferiores (tobillo) asociada con accidente cerebrovascular, cefaleas en migraña crónica, vejiga hiperactiva, hiperactividad neurogénica del detrusor o arrugas faciales del tercio superior.
CONTRAINDICACIONES
BOTOX® está contraindicado: En individuos con hipersensibilidad conocida a la toxina botulínica tipo A o a cualquiera de sus excipientes. En presencia de infección en el sitio (o sitios) de inyección propuesto(s).
BOTOX® para el tratamiento de hiperactividad del músculo detrusor de la vejiga, disfunción de la vejiga, está también contraindicado en:
- Pacientes con infección del tracto urinario.
- Pacientes con retención urinaria aguda quienes no se realizan rutinariamente limpieza con autocateterización intermitente (CIC).
USO EN EMBARAZO Y LACTANCIA
Embarazo: No hay datos adecuados sobre el uso de toxina botulínica tipo A en mujeres embarazadas. Estudios en animales han mostrado toxicidad reproductiva. Se desconoce el riesgo potencial para los seres humanos. BOTOX® no deberá ser utilizado durante el embarazo a menos que los beneficios superen claramente a los riesgos potenciales. Si se determina que el uso de BOTOX® es necesario durante el embarazo o la paciente se embaraza durante el tratamiento con BOTOX®, se le deberá advertir acerca de los riesgos potenciales.
Lactancia: No hay información que indique si BOTOX® es o no eliminado en la leche materna. No se recomienda utilizar BOTOX® durante la lactancia.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MAQUINARIA: Ha habido reportes de astenia, debilidad muscular, mareos y alteración visual después del tratamiento con BOTOX®, esto último puede hacer peligroso conducir o utilizar maquinaria.
REACCIONES ADVERSAS
General: En general, las reacciones adversas se presentan dentro de los primeros días posteriores a la inyección y, si bien suelen ser transitorias, pueden llegar a durar varios meses (o más tiempo en casos raros).
La debilidad muscular local representa la acción farmacológica esperada de la toxina botulínica en el tejido muscular. Sin embargo, se ha reportado debilidad de los músculos adyacentes y/o de músculos distantes del sitio de inyección.
Tal como cabe esperar para cualquier procedimiento de inyección, ha habido reportes de dolor localizado, inflamación, parestesia, hipoestesia, sensibilidad, hinchazón/edema, eritema, infección
localizada, sangrado y/o formación de hematomas en asociación con la inyección. El dolor y/o la ansiedad relacionados con la aguja han traído consigo respuestas vasovagales que han incluido hipotensión sintomática transitoria y síncope.
EXPERIENCIA DE ESTUDIOS CLINICOS
Reacciones adversas – frecuencia por indicación: A continuación se presenta la frecuencia de reacciones adversas documentada durante los estudios clínicos para cada indicación. La frecuencia es definida de la siguiente manera: muy comunes (=1/10), comunes (=1/100, <1/10), poco comunes (=1/1,000, <1/100) raros (=1/10,000, <1/1,000) y muy raros (<1/10,000).
Blefaroespasmo / espasmo hemifacial: Los datos de seguridad fueron recopilados a partir de estudios clínicos controlados y de estudios abiertos que incluyeron a 1732 pacientes tratados con BOTOX®. Se reportaron las siguientes reacciones adversas: Trastornos del sistema nervioso: Poco comunes: Mareos, parálisis facial; Trastornos oculares; Muy común: Ptosis del párpado; Comunes: Queratitis punteada, lagoftalmos, ojo
seco, fotofobia, irritación ocular, aumento del lagrimeo; Poco comunes: Queratitis, ectropión, diplopía, entropión, visión borrosa; Raros: Edema de los párpados; Muy raros: Queratitis ulcerativa, defecto epitelial corneal, perforación corneal. Trastornos de la piel y de los tejidos subcutáneos; Común: Equimosis; Poco común: Sarpullido. Trastornos generales y condiciones en el sitio de administración: Poco común: Fatiga.
Estrabismo: En los datos de seguridad recopilados a partir de estudios clínicos controlados, los cuales incluyeron a aproximadamente 2058 pacientes tratados con BOTOX®, se reportaron las siguientes reacciones adversas: Trastornos oculares: Muy comunes: Ptosis del párpado, trastorno del movimiento ocular; Poco comunes: Hemorragias retrobulbares oculares, penetración en el ojo, pupila de Holmes-Adie ; Raros: Hemorragia vítrea.
Distonía: Los datos de seguridad fueron recopilados a partir de un estudio doble ciego controlado con placebo que incluyó a 231 pacientes tratados con BOTOX®. Se reportaron las siguientes reacciones adversas: Infecciones e infestaciones: Comunes: Rinitis, infección del tracto respiratorio superior. Trastornos del sistema nervioso: Comunes: Mareos, hipertonía, hipoestesia, somnolencia, cefalea. Trastornos oculares; Poco comunes: Diplopía, ptosis del párpado. Trastornos respiratorios, torácicos y mediastínicos: Poco comunes: Disnea. Trastornos gastrointestinales: Muy común: Disfagia; Comunes: Boca seca, náuseas. Trastornos musculoesqueléticos y del tejido conjuntivo: Muy común: Debilidad muscular; Común: Rigidez musculoesquelética. Trastornos generales y condiciones en el sitio de administración: Muy común: Dolor; Comunes: Astenia, malestar, síndrome similar a resfriado; Poco
común: Pirexia
Parálisis cerebral pediátrica:
Espasticidad de las extremidades superiores: Se reportaron las siguientes reacciones adversas en 74 niños tratados para espasticidad de las extremidades superiores: Infecciones e infestaciones: Comunes: Influenza, neumonía. Trastornos del sistema nervioso: Comunes: Torpeza, hipoquinesia. Trastornos musculoesqueléticos y del tejido conjuntivo: Comunes: Debilidad muscular, espasmos musculares, dedo en gatillo. Trastornos renales y urinarios: Común: Poliuria. Trastornos gastrointestinales: Comunes: Vómito. Lesión, envenenamiento y complicaciones de procedimientos: Comunes: Dislocación de articulación, caída, contusión. Trastornos generales y condiciones en el sitio de administración: Muy común: Molestia en el sitio de inyección; Comunes: Formación de hematomas en el sitio de inyección, dolor en el sitio de inyección
Espasticidad de las extremidades inferiores: Se reportaron las siguientes reacciones adversas en los datos de seguridad recopilados a partir de dos estudios doble ciego, aleatorizados y controlados con placebo y de un estudio de extensión abierto, los cuales incluyeron a aproximadamente 304 pacientes tratados con BOTOX®: Infecciones e infestaciones: Muy comunes: Infección viral, infección del oído. Trastornos del sistema nervioso: Comunes: Somnolencia, alteración de la marcha, parestesia.
Trastornos de la piel y de los tejidos subcutáneos: Común: Sarpullido. Trastornos musculoesqueléticos y del tejido conjuntivo: Comunes: Mialgia, debilidad muscular, dolor en extremidad. Trastornos renales y urinarios: Común: Incontinencia urinaria. Lesión, envenenamiento y complicaciones de procedimientos: Comunes: Caída. Trastornos generales y condiciones en el sitio de administración: Comunes: Malestar, dolor en el sitio de inyección, astenia
Espasticidad focal de las extremidades superiores asociada con accidente cerebrovascular en adultos: Se reportaron las siguientes reacciones adversas en los datos de seguridad recopilados a partir de estudios doble ciego y abiertos que incluyeron a 339 pacientes tratados con BOTOX®: Trastornos del sistema nervioso: Común: Hipertonía; Poco comunes: Hipoestesia, cefalea, parestesia.
Trastornos vasculares: Poco comunes: Hipotensión ortostática. Trastornos gastrointestinales: Poco comunes: Náuseas. Trastornos de la piel y de los tejidos subcutáneos: Comunes: Equimosis; Poco comunes: Dermatitis, prurito, sarpullido. Trastornos musculoesqueléticos y del tejido conjuntivo: Comunes: Dolor en extremidad, debilidad muscular; Poco comunes: Artralgia, bursitis. Trastornos generales y condiciones en el sitio de administración: Comunes: Dolor en el sitio de inyección, pirexia,
enfermedad similar a influenza; Poco comunes: Astenia, dolor, hipersensibilidad en el sitio de inyección, malestar.
Espasticidad focal de las extremidades inferiores (tobillo) asociada con accidente cerebrovascular en adultos: Las siguientes reacciones adversas, se reportaron con mayor frecuencia (=1% ) en los pacientes tratados con BOTOX® y con mayor frecuencia que por pacientes tratados con placebo en ensayos clínicos doble ciego controlados por placebo para el tratamiento de la espasticidad de las extremidades inferiores en adultos. Trastornos generales y condiciones en el sitio de administración: Común: Edema periférico. Trastornos musculoesqueléticos y del tejido conjuntivo: Comunes: Artralgia, rigidez musculoesquelética. Trastornos de la piel y de los tejidos subcutáneos: Común: Sarpullido. No se observó cambio en el perfil de seguridad general con la administración dedosis repetidas.
Disfonía espasmódica: Se reportaron las siguientes reacciones adversas en los datos de seguridad recopilados a partir de los estudios clínicos con BOTOX®: Trastornos del sistema nervioso: Muy común: Disfonía. Trastornos respiratorios, torácicos y mediastínicos: Comunes: Aspiración, estridor; Poco comunes: Tos. Trastornos gastrointestinales: Muy común: Disfagia; Poco común: Náuseas. Trastornos generales y condiciones en el sitio de administración: Común: Dolor; Poco común: Enfermedad similar a influenza.
Hiperhidrosis: Hiperhidrosis axilar primaria: Se reportaron las siguientes reacciones adversas en los datos de seguridad recopilados a partir de estudios doble ciego y abiertos, los cuales incluyeron a 397 pacientes tratados con BOTOX®: Trastornos del sistema nervioso: Comunes: Cefalea, parestesia. Trastornos vasculares: Común: Sofocos. Trastornos gastrointestinales: Común: Náuseas. Trastornos de la piel y de los tejidos subcutáneos: Comunes: Hiperhidrosis, olor anormal de la piel, prurito, nódulo subcutáneo, alopecia. Trastornos musculoesqueléticos y del tejido conjuntivo: Común: Dolor en extremidad. Trastornos generales y condiciones en el sitio de administración: Muy común: Dolor en el sitio de inyección; Comunes: Dolor, edema en el sitio de inyección, hemorragia en el sitio de inyección, hipersensibilidad en el sitio de inyección, irritación en el sitio de inyección, astenia.
Nota: Se reportó un incremento de la sudoración no axilar en 4.5% de los pacientes, dentro del mes posterior a la inyección sin que se observara un patrón en lo que se refiere a los sitios anatómicos afectados. Se observó una resolución en aproximadamente 30% de los pacientes dentro de un periodo de cuatro meses.
En un estudio abierto de BOTOX® (50 U por axila) en pacientes adolescentes de 12 a 17 años de edad (N = 144), los eventos adversos más comunes (reportados en >3% de los pacientes) incluyeron amigdalitis (3.5%), nasofaringitis (4.9%) e infección del tracto respiratorio superior (21.5%). Las reacciones adversas de dolor en el sitio de inyección en hiperhidrosis fueron reportadas en dos pacientes cada uno. El perfil de seguridad de BOTOX® para el tratamiento de la hiperhidrosis en adolescentes fue similar al observado en la población adulta.
Hiperactividad del músculo detrusor de la vejiga:
Vejiga Hiperactiva: Las siguientes tasas fueron reportadas de los ensayos clínicos pivotales dobleciego, placebo-controlados, fase 3, durante el ciclo de tratamiento completo con BOTOX® 100 U:
Infecciones e infestaciones: Muy comunes: Infección del tracto urinario. Comunes: Bacteriuria. Trastornos renales y urinarios Muy comunes: Disuria. Comunes: Retención urinaria, volumen de orina residual*, polaquiuria.
*PVR elevado no requiere cateterización.
Las reacciones adversas relacionadas al procedimiento que se presentaron con una frecuencia común fueron disuria y hematuria.
La cateterización fue iniciada en el 6.5% después del tratamiento con BOTOX 100 Unidades en comparación con 0.4% en el grupo placebo.
No se observaron cambios en el perfil general de seguridad con dosis repetidas.
Hiperactividad Neurogénica del Detrusor: Se reportaron las siguientes tasas en los estudios doble ciego sobre BOTOX® (200 U) durante el ciclo de tratamiento completo (duración media de la exposición: 44 semanas): Infecciones e infestaciones: Muy común: Infección del tracto urinario. Trastornos psiquiátricos: Común: Insomnio. Trastornos gastrointestinales: Común: Estreñimiento. Trastornos musculoesqueléticos y del tejido conjuntivo: Comunes: Debilidad muscular, espasmo muscular. Trastornos renales y urinarios: Muy común: Retención urinaria; Comunes: Hematuria*, disuria*, divertículo de la vejiga. Trastornos generales y condiciones en el sitio de administración: Comunes: Fatiga, alteración de la marcha. Lesión, envenenamiento y complicaciones de procedimientos: Comunes: Disreflexia autonómica*, caída. * Reacciones adversas relacionadas con los procedimientos
No se observó un cambio del perfil de seguridad general con la administración de dosis repetidas.
No se observó una diferencia de la tasa anual de exacerbaciones de MS (número de eventos de exacerbación de MS por año-paciente) (BOTOX® = 0.23, placebo = 0.20) en los pacientes con MS enrolados en los estudios esenciales.
De los pacientes sin cateterización en la línea basal antes del tratamiento, se inició cateterización en 38.9% después del tratamiento con BOTOX® (200 U) en comparación con 17.3% con el placebo.
Alternativo en la Profilaxis del Dolor de Cabeza en Migraña Crónica: Los datos de seguridad fueron recopilados a partir de dos estudios doble ciego controlados con placebo que incluyeron a 687 pacientes tratados con BOTOX®. Se reportaron las siguientes
reacciones adversas: Trastornos del sistema nervioso: Comunes: Cefalea, migraña, paresia facial. Trastornos oculares: Comunes: Ptosis del párpado. Trastornos gastrointestinales: Poco comunes: Disfagia. Trastornos generales y condiciones en el sitio de administración: Comunes: Dolor en el sitio de inyección. Trastornos de la piel y de los tejidos subcutáneos: Comunes: Prurito, salpullido; Poco comunes: Dolor de la piel. Trastornos musculoesqueléticos y del tejido conjuntivo: Comunes: Dolor del cuello, rigidez musculoesquelética, debilidad muscular, mialgia, dolor musculoesquelético, espasmos musculares, tirantez muscular; Poco comunes: Dolor en la mandíbula.
Se reportó migraña (incluyendo empeoramiento de migraña) en 3.8% de los pacientes tratados con BOTOX® y 2.6% de quienes recibieron un placebo, típicamente dentro del primer mes posterior al tratamiento. Estas reacciones no volvieron a presentarse en forma constante con los ciclos de tratamiento subsecuentes y la incidencia general disminuyó con las repeticiones del tratamiento. La tasa de discontinuaciones debidas a eventos adversos en estos estudios de Fase 3 fue de 3.8% con BOTOX® vs. 1.2% con placebo.
Líneas faciales Hiperfuncionales: Líneas glabelares: Se reportaron los siguientes eventos adversos en los datos de seguridad recopilados a partir de dos estudios doble ciego, multicéntricos y controlados con placebo que incluyeron a 405 pacientes tratados con BOTOX®: Trastornos del sistema nervioso: Comunes: Cefalea, parestesia, paresis facial. Trastornos oculares: Común: Ptosis del párpado. Trastornos gastrointestinales: Común: Náuseas: Trastornos de la piel y de los tejidos subcutáneos: Comunes: Eritema, tirantez de la piel. Trastornos musculoesqueléticos y del tejido conjuntivo: Común: Debilidad muscular. Trastornos generales y condiciones en el sitio de administración: Comunes: Dolor facial, edema en el sitio de inyección, equimosis, dolor en el sitio de inyección, irritación en el sitio de inyección
Frente: Se reportaron las siguientes reacciones adversas en los datos de seguridad recopilados a partir de los estudios clínicos en pacientes tratados con BOTOX®:
Trastornos del sistema nervioso: Muy común: Cefalea. Trastornos oculares: Muy común: Edema de los párpados. Trastornos generales y condiciones en el sitio de administración: Muy comunes: Formación de hematomas en el sitio de inyección, prurito en el sitio de aplicación, dolor facial
Canto lateral (patas de gallo): Se reportaron las siguientes reacciones adversas en los datos de seguridad recopilados a partir de los estudios clínicos en pacientes tratados con BOTOX®: Trastornos del sistema nervioso: Comunes: Cefalea : Trastornos oculares: Comunes: Ptosis del párpado, edema del párpado; Raros: Diplopía. Trastornos musculoesqueléticos y del tejido conjuntivo: Raros: Debilidad muscular. Trastornos generales y condiciones en el sitio de administración: Muy común: Formación de hematomas en el sitio de inyección; Comunes: Dolor facial No se reportaron eventos adversos con la administración simultánea de 44U para el tratamiento de las líneas de patas de gallo y líneas glabelares.
EXPERIENCIA POST-MARKETING
Después del tratamiento con BOTOX®, ha habido raros reportes espontáneos de muerte, algunos asociados con disfagia, neumonía, y/o otras debilidades significativas.
Reacciones graves y/o inmediatas de hipersensibilidad como, anafilaxis y enfermedad del suero, han sido reportadas en raras ocasiones, como también lo han sido otras manifestaciones de hipersensibilidad incluyendo urticaria, edema de tejidos blandos y disnea. Algunas de estas reacciones han sido reportadas después del uso de BOTOX® ya sea por sí solo o en conjunción con otros productos asociados con reacciones similares. Se reportó un caso fatal de anafilaxis en el cual el paciente murió después de que se le inyectara BOTOX® diluido inapropiadamente en 5 mL de lidocaína al 1%. El papel causal de BOTOX®, de la lidocaína o de ambos no ha sido determinado.
Ha habido reportes raros de eventos adversos relacionados con el sistema cardiovascular (incluyendo arritmia e infarto del miocardio, algunos de ellos con desenlace fatal) después del tratamiento con BOTOX®. Algunos de los pacientes en cuestión presentaban factores de riesgo incluyendo enfermedad cardiovascular.
También se han reportado crisis convulsivas de nueva aparición o recurrentes después del tratamiento con BOTOX®, típicamente en pacientes con predisposición para sufrir estos eventos.
Se ha reportado glaucoma por cierre angular en muy raras ocasiones después del tratamiento con BOTOX® para blefaroespasmo.
Se ha reportado lagoftalmos después de la inyección con BOTOX® en las líneas glabelares o en las patas de gallo.
La siguiente lista incluye reacciones medicamentosas adversas u otros eventos adversos médicamente relevantes que han sido reportados desde que el fármaco fue comercializado: denervación/atrofia muscular, depresión respiratoria y/o insuficiencia respiratoria, disnea y neumonía por aspiración, disartria, boca seca, estrabismo, neuropatía periférica, dolor abdominal, diarrea, náuseas, vómito, pirexia, anorexia, visión borrosa, alteración visual, hipoacusia, tinnitus, vértigo, parálisis facial, paresia
facial, plexopatía braquial, radiculopatía, síncope, hipoestesia, malestar, mialgia, miastenia grave, parestesia, sarpullido, eritema multiforme, prurito, dermatitis psoriasiforme, hiperhidrosis y alopecia incluyendo madarosis
INTERACCIONES MEDICAMENTOSAS
En teoría, el efecto de la toxina botulínica tipo A podría ser potenciado por los antibióticos aminoglucósidos o por medicamentos que interfieren con la transmisión neuromuscular (p. ej., agentes bloqueadores neuromusculares). No se han realizado pruebas específicas para establecer la posibilidad de interacción clínica con otros productos medicamentosos. No se han reportado interacciones medicamentosas de significancia clínica.
El efecto de la administración de diferentes serotipos de neurotoxina botulínica al mismo tiempo o con pocos meses de separación entre ellas es desconocido. Es posible que la debilidad excesiva se vea potenciada por la administración de una toxina botulínica antes de que los efectos de otra toxina botulínica administrada previamente se hayan resuelto.
ADVERTENCIAS Y PRECAUCIONES
General: La anatomía relevante y cualesquiera alteraciones de la anatomía debidas a procedimientos quirúrgicos previos deben ser ponderadas antes de administrar BOTOX®. Debe evitarse la inyección de BOTOX® en estructuras anatómicas vulnerables.
Eventos adversos serios incluyendo resultados fatales han sido reportados en pacientes que habían recibido tratamiento con BOTOX® inyectado directamente en las glándulas salivares, la región orolingual- faríngea, esófago y estómago. Algunos pacientes tenían preexistencia de disfagia o debilidad significativa.
Se ha reportado neumotórax asociado con el procedimiento de inyección después de la administración de BOTOX® cerca del tórax. Es necesario tener precaución al inyectarlo cerca del pulmón, particularmente de los ápices.
Se deberá tener precaución al usar BOTOX® en presencia de inflamación en el sitio (o sitios) de inyección propuesto(s) o cuando haya debilidad excesiva o atrofia en el músculo objetivo.
Como se espera con cualquier procedimiento de inyección, Ha habido reportes de dolor localizado, inflamación, parestesia, hipoestesia, sensibilidad, hinchazón/edema, eritema, infección localizada, sangrado y/o formación de hematomas en asociación con la inyección. El dolor y/o la ansiedad relacionados con la aguja han traído consigo respuestas vasovagales que han incluido hipotensión sintomática transitoria y síncope.
Reacciones de Hipersensibilidad: Reacciones serias y/o inmediatas de hipersensibilidad tales como, anafilaxis y enfermedad del suero han sido reportadas en raras ocasiones, como también lo han sido otras manifestaciones de hipersensibilidad incluyendo urticaria, edema de tejidos blandos y disnea. Algunas de estas reacciones han sido reportadas después del uso de BOTOX® ya sea por sí solo o en conjunción con otros productos asociados con reacciones similares. De presentarse una reacción de esta naturaleza, no se deberán administrar más inyecciones y se deberá instituir una terapia médica apropiada en forma inmediata. Se reportó un caso fatal de anafilaxis en el cual el paciente murió después de que se le inyectara BOTOX® diluido inapropiadamente en 5 mL de lidocaína al 1%. La causalidad de BOTOX®, de la lidocaína o de ambos no ha sido determinado.
Trastornos Neurológicos Preexistentes: Se deberá tener precaución extrema al administrar BOTOX® a individuos con enfermedades neuropáticas motoras periféricas (p. ej., esclerosis lateral amiotrófica o neuropatía motora) o con trastornos de la conexión neuromuscular (p. ej., miastenia grave o síndrome de Lambert-Eaton). Los pacientes con trastornos de la conexión neuromuscular pueden llegar a presentar un incremento del riesgo de efectos sistémicos clínicamente significativos (incluyendo disfagia severa y afectación respiratoria), al recibir dosis típicas de BOTOX®. Ha habido casos raros de administración de toxina botulínica a pacientes con trastornos, conocidos o no conocidos, de la conjunción neuromuscular, en los cuales los pacientes han mostrado sensibilidad extrema a los efectos sistémicos de las dosis clínicas típicas. En algunos de estos casos, la disfagia ha durado varios meses y ha hecho necesario colocar un tubo de alimentación gástrica. Al ser expuestos a dosis sumamente elevadas, los pacientes con trastornos neurológicos (p. ej., parálisis cerebral pediátrica o espasticidad del adulto) también pueden llegar a presentar un aumento del riesgo de efectos sistémicos clínicamente significativos.
Efectos adversos distantes del sitio de inyección: Datos de seguridad obtenidos después de la comercialización para BOTOX® y otras toxinas botulínicas aprobadas sugieren que, en ciertos casos, es posible que los efectos de la toxina sean observados en lugares distantes del sitio de inyección. Los síntomas concordantes con el mecanismo de acción de la toxina botulínica han sido reportados horas a semanas después de la inyección y han incluido debilidad muscular, ptosis, diplopía, visión borrosa, debilidad facial, disfagia, trastornos del habla y de la deglución, estreñimiento, neumonía por aspiración, dificultad para respirar y depresión respiratoria. Es probable que el riesgo de presentar síntomas sea mayor en los niños tratados para espasticidad, pero los síntomas también pueden presentarse en pacientes con condiciones y comorbilidades subyacentes que los predispondrían a padecer dichos síntomas, incluyendo a adultos tratados para espasticidad y otras condiciones, y en pacientes que son tratados con dosis elevadas. La dificultad para deglutir y para respirar pueden amenazar la vida y se ha reportado la muerte, aunque no se ha establecido una asociación causal definitiva con BOTOX®.
Se deberá aconsejar a los pacientes o cuidadores que busquen atención médica inmediata de surgir trastornos de la deglución, del habla o de tipo respiratorio.
Sistema Cardiovascular: Ha habido reportes de eventos adversos que han involucrado al sistema cardiovascular después de la administración de BOTOX®, incluyendo arritmia e infarto del miocardio, algunos de ellos con desenlace fatal. Algunos de los pacientes en cuestión presentaban factores de riesgo incluyendo enfermedad cardiovascular preexistente. Se desconoce la relación exacta de estos eventos con BOTOX®.
Crisis Convulsivas: Se han reportado crisis convulsivas de nueva aparición o recurrentes, típicamente en pacientes con predisposición para presentar estos eventos. La relación exacta entre estos eventos y la inyección con BOTOX® no ha sido establecida. Los reportes en niños correspondieron predominantemente a pacientes con parálisis cerebral tratados para espasticidad.
Inmunogenicidad: La formación de anticuerpos neutralizantes de la toxina botulínica tipo A pueden reducir la efectividad del tratamiento con BOTOX® al inactivar la actividad biológica de la toxina. Los factores críticos para la formación de anticuerpos neutralizantes no han sido bien caracterizados. El potencial de formación de anticuerpos se puede minimizar inyectando las dosis efectivas más bajas, administradas con el mayor intervalo posible entre inyecciones.
Albúmina Humana: Este producto contiene albúmina sérica humana, un derivado de la sangre humana. Con base en procesos eficaces de selección de donantes y manufactura del producto, éste conlleva un riesgo
extremadamente remoto de transmisión de enfermedades virales. El riesgo teórico de transmisión de enfermedad de Creutzfeldt-Jakob (CJD) también es considerado como extremadamente remoto. No se han identificado casos de transmisión de enfermedades virales o CJD en asociación con la albúmina.
Blefaroespasmo/ Espasmo hemifacial: La reducción del parpadeo después de la inyección de BOTOX® en el músculo orbicular del ojo puede traer consigo exposición de la córnea, defectos epiteliales persistentes y ulceración de la córnea, especialmente en pacientes con trastornos del nervio craneal VII. Se ha presentado un caso de perforación de la córnea en un ojo afáquico que hizo necesario un injerto de córnea debido a este efecto. Se deberá efectuar una evaluación cuidadosa de la sensación de la córnea en los ojos previamente operados y evitar la inyección en el área del párpado medial inferior para evitar ectropión, así como tratar eficazmente todo defecto epitelial. Puede que esto haga necesario el uso de gotas protectoras, ungüentos o lentes de contacto blandos terapéuticos o el cierre del ojo con un parche u otros medios.
Debido a la actividad anticolinérgica de la toxina botulínica, se deberá tener precaución al tratar a pacientes con riesgo de glaucoma por cierre angular, incluyendo a pacientes con ángulos anatómicamente estrechos.
Estrabismo: BOTOX® no es eficaz para el estrabismo paralítico crónico excepto para reducir la contractura del antagonista en conjunción con reparación quirúrgica. La eficacia de BOTOX® en desviaciones de más de 50 dioptrías de prisma, en el estrabismo restrictivo, en el síndrome de Duane con debilidad del recto lateral y en el estrabismo secundario causado por recesión quirúrgica excesiva previa del antagonista es dudosa. Puede que para aumentar la eficacia sea necesario realizar múltiples inyecciones a través del tiempo.
Durante la administración de BOTOX® para el tratamiento del estrabismo se han presentado hemorragias retrobulbares suficientes para afectar la circulación retinal, como consecuencia de penetraciones de la aguja en la órbita. Se recomienda tener acceso a instrumentos apropiados para examinar y descomprimir la órbita. También se han presentado penetraciones del globo ocular por las agujas. Deberá disponerse de un oftalmoscopio para diagnosticar esta condición.
Puede que la inducción de parálisis en uno o más músculos extraoculares produzca desorientación espacial, visión doble o señalización pasada (“past-pointing”). Cubrir el ojo el ojo afectado puede aliviar estos síntomas.
Distonía Cervical: La disfagia es un evento adverso reportado comúnmente después del tratamiento de pacientes con distonía cervical, con todos los tipos de toxinas botulínicas. Se deberá informar a los pacientes con distonía cervical sobre la posibilidad de experimentar disfagia que puede ser leve, pero podría ser severa. Puede que la disfagia persista por dos a tres semanas después de la inyección, pero se ha reportado una duración de hasta 5 meses después de la inyección. Como consecuencia de la disfagia existe el potencial de que se presente aspiración, disnea y, ocasionalmente, la necesidad de alimentación con tubo. En casos raros se ha reportado disfagia seguida de neumonía por aspiración y muerte.
Las inyecciones en el elevador de la escápula pueden asociarse con un aumento del riesgo de infección de las vías respiratorias superiores y disfagia.
La disfagia ha contribuido a una disminución de la ingesta de alimento y agua que ha traído consigo pérdida de peso y deshidratación. Puede que los pacientes con disfagia subclínica presenten un mayor riesgo de experimentar disfagia más severa después de una inyección de BOTOX®.
Limitar la dosis inyectada en ambos músculos esternocleidomastoideos a menos de 100 U puede reducir la incidencia de disfagia. Se ha reportado que los pacientes con menor masa muscular en el cuello o los pacientes que reciben inyecciones bilaterales en los esternocleidomastoideos presentan mayor riesgo de disfagia. La disfagia es atribuible a la difusión localizada de la toxina a la musculatura esofágica.
Se deberá aconsejar a los pacientes o cuidadores que busquen atención médica inmediata de surgir trastornos de la deglución, del habla o de tipo respiratorio.
Espasticidad focal asociada con parálisis cerebral pediátrica y espasticidad focal de las extremidades superiores e inferiores (tobillo) asociada con accidente cerebrovascular en adultos: BOTOX® es un tratamiento para espasticidad focal que sólo ha sido estudiado en asociación con regímenes de atención estándar usuales y no tiene por propósito reemplazar dichas opciones de tratamiento. No es probable que BOTOX® sea eficaz para mejorar el rango de movimiento en una articulación afectada por una contractura fija.
No se debe usar BOTOX® para el tratamiento de la espasticidad focal de las extremidades inferiores en pacientes adultos que han sufrido un accidente cerebrovascular si no se espera que la reducción en el tono muscular produzca una función mejorada (por ejemplo, mejora en la marcha), o mejoría de los síntomas (por ejemplo, reducción del dolor) o facilite los cuidados que se le prestan al paciente.
Se debe proceder con cautela cuando se tratan pacientes con espasticidad secundaria a un accidente cerebrovascular los cuales pueden correr un mayor riesgo de sufrir caídas.
Se debe proceder con cautela cuando se usa BOTOX® para el tratamiento de la espasticidad focal en las extremidades inferiores en pacientes geriátricos que han sufrido un accidente cerebrovascular con co-morbilidades significativas y solo se debe iniciar el tratamiento si se considera que los beneficios superan los potenciales riesgos.
Ha habido reportes espontáneos raros de muerte, en ocasiones asociada con neumonía por aspiración, en niños con parálisis cerebral severa después del tratamiento con toxina botulínica. Se
deberá tener precaución al tratar a pacientes pediátricos que presentan debilidad neurológica significativa, disfagia o un historial reciente de neumonía por aspiración o enfermedad pulmonar.
Disfonía espasmódica: En general, el tratamiento de la disfonía espasmódica con BOTOX® deberá ser evitado en pacientes que habrán de someterse a una cirugía electiva que requerirá anestesia general, ya que BOTOX® relaja las cuerdas vocales, lo cual potencialmente incrementa el riesgo de aspiración perioperatoria,
etc. Se recomienda que este procedimiento sea llevado a cabo por médicos apropiadamente capacitados y en instalaciones preparadas para manejar el estridor reflejo que se llegara a presentar asociado con el procedimiento.
Hiperhidrosis:
Hiperhidrosis primaria de las axilas: Los pacientes deberán ser evaluados para detectar potenciales causas de hiperhidrosis secundaria (p. ej., hipertiroidismo o feocromocitoma) a fin de evitar el
tratamiento sintomático de la hiperhidrosis sin diagnóstico y/o tratamiento de la enfermedad subyacente.
Hiperactividad del músculo detrusor de la vejiga:
Disfunción de la vejiga
Se debe tener las precauciones médicas adecuadas al realizar la citoscopia.
En pacientes que no están cateterizados, el volumen de orina residual después de evacuar debe evaluarse 2 semanas después del tratamiento y periódicamente como es médicamente apropiado hasta 12 semanas. A los pacientes se les debe instruir para que contacten su médico en caso de experimentar dificultades al evacuar ya que puede ser necesaria la cateterización.
Debido al riesgo de retención urinaria, solamente deben ser considerados para el tratamiento los pacientes que estén dispuestos o sean aptos para iniciar cateterización post-tratamiento, en caso que se requiera.
Hiperactividad Neurogénica del Detrusor
En estos pacientes, se puede presentar disreflexia autonómica asociado con el procedimiento, la cual podría hacer necesaria una terapia médica pronta.
Líneas Faciales Hiperfuncionales:
Arrugas en la parte superior del rostro – frente, canto lateral y glabelares: La reducción del parpadeo causada por la inyección de BOTOX® en el músculo orbicular del ojo puede ocasionar exposición de la córnea, defectos epiteliales persistentes y ulceración de la córnea, especialmente en pacientes con trastornos del nervio craneal VII. Se deberá tener precaución al utilizar tratamiento con BOTOX® en pacientes con inflamación en el sitio de inyección, asimetría facial pronunciada, ptosis, dermatocalasis excesiva, cicatrices dérmicas profundas o piel sebácea gruesa, o imposibilidad de disminuir sustancialmente las líneas glabelares extendiéndolas físicamente.
Alternativo en la Profilaxis del Dolor de Cabeza en Migraña Crónica: Consulte las Advertencias y Precauciones precedentes, las cuales incluyen información específica para las inyecciones en cabeza y cuello, ya que los sitios de inyección son similares.
INSTRUCCIONES DE USO, MANEJO Y DISPOSICIÓN:
Como buena práctica, se recomienda reconstituir el vial y preparar la jeringa sobre toallas de papel con un recubrimiento de plástico por si hubiese derrame. BOTOX® debe ser reconstituido solamente con solución salina libre de preservantes (Cloruro de sodio al 0.9% para inyección). Extraiga una cantidad apropiada de diluyente, según tabla a continuación, a una jeringa. Para la técnica de reconstitución de las inyecciones intradetrusor en el tratamiento de hiperactividad neurogénica del detrusor, refiérase a la posología y forma de administración para esta indicación.
|
Tabla de dilución para Viales de BOTOX® BTX-A® 50 U BOTOX® 100 U y BOTOX® 200 U |
|||
|
Diluyente adicionado (cloruro de sodio al 0.9% para inyección) |
Vial de 50 U |
Vial de 200 U |
Vial de 100 U |
|
Dosis resultante/ unidades por 0.1 mL |
Dosis resultante/ unidades por 0.1 mL |
Dosis resultante/ unidades por 0.1 mL |
|
|
0.5 mL |
10 |
40 |
20 |
|
1 mL |
5 |
20 |
10 |
|
2 mL |
2.5 |
10 |
5 |
|
4 mL |
1.25 |
5 |
2.5 |
|
8 mL |
NA |
2.5 |
1.25 |
|
10 mL |
NA |
2 |
1 |
|
Nota: Estas diluciones están calculadas para un volumen de inyección de 0.1 mL. También es posible un aumento o disminución de la dosis de BOTOX® administrando un volumen menor o mayor de inyección desde 0.05 mL (50% de disminución de la dosis) a 0.15 mL (50% de aumento de la dosis). |
|||
Inyecte el diluyente lentamente dentro de la vial. Deseche el vial si se evidencia pérdida del vacío (es decir, si el diluyente no es succionado hacia el vial al introducir la aguja). BOTOX® reconstituido es una solución transparente incolora a ligeramente amarillenta libre de material particulado. Antes de utilizar la solución reconstituida, la misma debe ser inspeccionada visualmente para verificar su transparencia y ausencia de partículas. El producto debe ser utilizado una sola vez y la solución sobrante debe ser desechada.
Todos los viales, incluyendo los que hayan vencido y el equipo utilizado con el medicamento, deben desecharse cuidadosamente como se hace con todo desecho médico. En caso que se requiera la desactivación de la toxina (p.e. derrames), se recomienda el uso de solución de hipoclorito diluída (0.5% o 1%) durante 5 minutos antes de ser descartado como desecho médico.
POSOLOGÍA Y MÉTODO DE ADMINISTRACIÓN
Administración: BOTOX® está indicado para uso intramuscular, intradérmico o intradetrusor de acuerdo al uso indicado
General
BOTOX® debe ser administrado solo por médicos con la calificación apropiada y con experiencia en el tratamiento y en el uso del equipo necesario.
Los niveles de dosis óptimos y el número de sitios de inyección por músculo no han sido establecidos para todas las indicaciones. La dosis exacta y el número de sitios de inyección deberán ser determinados de acuerdo con las necesidades del paciente con base en el tamaño, el número y la ubicación de los músculos involucrados, en la severidad de la enfermedad, en la presencia de debilidad muscular local, en la respuesta al tratamiento previo y en la condición médica del paciente.
Como con cualquier tratamiento con medicamento, la administración inicial en un paciente sin experiencia previa al tratamiento deberá comenzar con la dosis más baja recomendada. Si fuese necesario, dicha dosis puede ser incrementada gradualmente en tratamientos subsecuentes hasta alcanzar la dosis máxima generalmente estudiada o indicada.
En general, BOTOX® no deberá ser inyectado con una frecuencia mayor a un tratamiento cada tres meses. Se deben seguir las indicaciones específicas con respecto a la dosis y a la administración. Si bien no hay datos disponibles derivados de estudios clínicos controlados acerca del tratamiento concurrente de múltiples indicaciones, en general, como consideración práctica al tratar a pacientes adultos (incluyendo el tratamiento para múltiples indicaciones), la dosis acumulativa máxima no deberá exceder 400 U en un intervalo de 3 meses. En el tratamiento de paciente pediátricos, la dosis acumulativa máxima en un intervalo de 3 meses, generalmente no deberá exceder 8 U/kg de peso corporal o 300 U, eligiendo el menor de los dos valores. Los resultados clínicos (incluyendo los riesgos) para dosis más elevadas en los diferentes grupos de edad no han sido establecidos completamente.
El término “Unidad” (U) en el cual se basa la dosis es una medición específica de la actividad de la toxina que es propia de la formulación de toxina botulínica tipo A de Allergan. Por lo tanto, las U utilizadas para describir la actividad de BOTOX® son diferentes de las utilizadas para describir la actividad de otras preparaciones de toxina botulínica y las U representativas de la actividad de BOTOX®, no son intercambiables con las U de otros productos.
Las dosis para los pacientes mayores de 65 años son las mismas que para los adultos más jóvenes. La administración inicial deberá comenzar con la dosis más baja recomendada para la indicación específica. (Ver sección 11)
La seguridad y eficacia de BOTOX® no ha sido establecida en niños menores de 2 años, para la indicación de espasticidad de las extremidades superiores e inferiores asociada con parálisis cerebral; en niños menores de 12 años para las indicaciones de blefaroespasmo, espasmo hemifacial, estrabismo, disfonía espasmódica o hiperhidrosis, en pacientes menores de 16 años para la indicación de distonía cervical ni en pacientes menores de 18 años para las indicaciones de espasticidad de las extremidades superiores e inferiores (tobillo) asociada con accidente cerebrovascular, cefaleas en migraña crónica, vejiga hiperactiva, hiperactividad neurogénica del detrusor o arrugas en la parte superior del rostro.
En investigaciones clínicas, BOTOX® reconstituido ha sido inyectado utilizando una aguja estéril calibre 25 a 30, de longitud apropiada para el músculo esquelético y para las indicaciones dermatológicas. La localización del músculo objetivo mediante guía electromiográfica, estimulación del nervio o técnicas ecográficas pueden ser útil. La inyecciones intradetrusor son realizadas bajo visualización directa vía cistoscopio con una aguja apropiada.
Se recomienda que BOTOX® sea usado para un único uso y en una única sesión de tratamiento. Para instrucciones más específicas sobre la dilución, manejo y disposición de residuos del producto ver sección 16 Instrucciones de Uso, Manejo y Disposición.
Blefaroespasmo: La dosis recomendada inicial es de 1.25 a 2.5 U (volumen de 0.05 mL a 0.1 mL en cada sitio) inyectadas en el orbicular medial y lateral del párpado superior y en el orbicular lateral del párpado
inferior.
Puede que el evitar la inyección cerca del elevador palpebral superior puede reduzca la incidencia de ptosis del párpado. El evitar la inyección en el párpado inferior medial (reduciendo así la difusión al oblicuo inferior) reduzca la incidencia de diplopía. Puede presentarse con frecuencia equimosis en los tejidos blandos de los párpados. Ello puede ser minimizado aplicando presión ligera al sitio de inyección inmediatamente después de la administración.
En general, el efecto inicial de las inyecciones es observado dentro de 3 días y el efecto pico es alcanzado una a dos semanas después del tratamiento. Cada tratamiento dura aproximadamente 3 meses, una vez transcurridos los cuales, el procedimiento puede ser repetido según sea necesario.
La dosis inicial no deberá exceder 25 U por ojo. En las sesiones de tratamiento sucesivas, la dosis puede ser incrementada al doble en comparación con la dosis administrada previamente, si se considera que la respuesta al tratamiento inicial fue insuficiente (definida como un efecto que dura menos de dos meses). Sin embargo en la mayoría de las situaciones, parece haber un aumento mínimo del beneficio al inyectar más de 5 U por sitio.
En general, la dosis acumulativa de BOTOX® para el tratamiento del blefaroespasmo no deberá exceder 200 U en un periodo de 2 meses.
Espasmo Hemifacial: Los pacientes con espasmo hemifacial o trastornos del nervio craneal VII deberán ser tratados como los pacientes con blefaroespasmo unilateral, inyectándose otros músculos faciales afectados (corrugador, cigomático mayor, orbicular de la boca) según sea necesario.
En general, la dosis acumulativa de BOTOX® para el tratamiento de espasmo hemifacial no deberá exceder 200 U en un periodo de 2 meses.
Estrabismo: BOTOX® debe ser inyectado, en los músculos extraoculares, siendo necesaria orientación electromiográfica. Para preparar el ojo para una inyección de BOTOX®, se recomienda la administración de varias gotas de un anestésico local y un descongestionante ocular varios minutos antes de la inyección.
Dosis iniciales: utilícense las dosis más bajas para el tratamiento de desviaciones leves y dosis más elevadas para desviaciones más pronunciadas.
1. Para músculos verticales y para estrabismo horizontal de menos de 20 dioptrías de prisma: 1.25 a 2.5 U (0.05 a 0.10 mL) en cualquier músculo individual dado.
2. Para estrabismo horizontal de 20 a 50 dioptrías de prisma: 2.5 a 5 U (0.10 a 0.20 mL) en cualquier músculo individual dado.
3. Para parálisis del nervio craneal VI que persiste durante un mes o más: 1.25 a 2.5 U (0.05 a 0.10 mL) en el recto medial.
Las dosis iniciales de BOTOX® suelen inducir parálisis de los músculos inyectados uno a dos días después de la inyección. La intensidad de la parálisis se incrementa durante la primera semana. La parálisis dura 2 a 6 semanas y se resuelve gradualmente a lo largo de un periodo similar. Las correcciones excesivas de más de 6 meses de duración han sido raras.
Aproximadamente la mitad de los pacientes tratados necesitará dosis adicionales debido a una respuesta clínica inadecuada del músculo después de la dosis inicial o debido a factores mecánicos tales como restricciones o desviaciones altas, o a la falta de fusión motora binocular para estabilizar la alineación.
Se recomienda que los pacientes sean valorados 7-14 días después de cada inyección para evaluar el efecto de la dosis aplicada.
Las dosis subsecuentes para los pacientes que experimenten parálisis completa del músculo objetivo deberán ser comparables a la dosis inicial. Las dosis subsecuentes para los pacientes que experimenten parálisis incompleta del músculo objetivo pueden ser incrementadas hasta dos veces, en comparación con la dosis administrada previamente. No se deberán administrar nuevas inyecciones hasta que los efectos de la dosis anterior hayan desaparecido, como lo evidencia el retorno de la función del músculo inyectado y de los músculos adyacentes.
La dosis máxima recomendada en forma de una inyección única para cualquier músculo individual determinado es de 25 U. El volumen recomendado de inyección de BOTOX® para el tratamiento del estrabismo es de 0.05 mL a 0.15 mL por músculo.
Distonia Cervical: El tratamiento de la distonía cervical puede incluir, aunque sin limitarse a, la inyección de BOTOX® en el esternocleidomastoideo, el elevador de la escápula, los escalenos, el esplenio de la cabeza, el semiespinal, el largo y/o el trapecio o trapecios. En caso de haber cualquier dificultad para aislar los músculos individuales, las inyecciones deberán ser realizadas por un médico experimentado empleando asistencia electromiográfica.
En un estudio clínico controlado, las dosis variaron entre 95 y 360 U (con una media aproximada de 240 U). Como es el caso con cualquier tratamiento con un fármaco, la dosis inicial en un paciente sin experiencia previa al tratamiento deberá comenzar con la dosis más baja recomendada. No se deberán administrar más de 50 U en un sitio individual determinado. Puede que el limitar la dosis total inyectada en los músculos esternocleidomastoideos a 100 U o menos reduzca la incidencia de disfagia. El número óptimo de sitios de inyección depende del tamaño del músculo.
Por lo general, la mejoría clínica suele tener lugar dentro de las primeras dos semanas después de la inyección. El beneficio clínico máximo suele presentarse antes de que transcurran seis semanas después de la inyección. No se recomienda que los intervalos de tratamiento sean menores a dos meses. La duración del efecto benéfico reportada en estudios clínicos ha mostrado una variación sustancial (de 2 a 32 semanas) y típicamente ha sido de 12 a 16 semanas, aproximadamente.
En general, la dosis acumulativa máxima para distonía cervical no deberá exceder 360 U en un intervalo de 3 meses.
Espasticidad focal en niños de >= 2 años de edad: Antes de la inyección de BOTOX® deberá realizarse identificación de los objetivos del tratamiento y de los músculos específicos responsables del patrón limitante de espasticidad. Es necesario un examen clínico para evaluar a los músculos en un patrón de espasticidad focal y es posible que el uso de electromiografía, ultrasonido muscular o estimulación eléctrica aumente la precisión de las inyecciones de BOTOX®.
En pacientes pediátricos, la máxima dosis acumulativa en un intervalo de 3 meses por lo general no debe exceder 8.0 U/kg de peso corporal o 300 U, eligiendo el menor de los dos valores.
En estudios clínicos para el tratamiento de la espasticidad de las extremidades superiores, la dosis por músculo varió entre 0.5 y 2.0 U/kg de peso corporal en las extremidades superiores por sesión de tratamiento. La dosis total varió entre 3.0 y 8.0 U/kg de peso corporal y no excedió de 300 Unidades dividido entre los músculos seleccionados en cualquier sesión de tratamiento.
En estudios clínicos para el tratamiento de la deformidad en pie equino, la dosis por músculo varió de 2.0 a 4.0 Unidades/kg de peso corporal en las extremidades inferiores por sesión de tratamiento. La dosis total fue de 4.0 U/kg de peso corporal ó 200 Unidades (la que fuera menor) dividida entre uno o dos sitios en el músculo gastrocnemio medial y lateral de una o las dos piernas en cualquier sesión de tratamiento. Puede que, después de la inyección inicial en el músculo gastrocnemio, sea necesario
considerar incluir al tibial anterior o al tibial posterior para una mejoría adicional de la posición del pie al golpear el talón y al permanecer de pie.
La siguiente tabla busca suministrar lineamientos de dosificación para la inyección de BOTOX® en el tratamiento de la espasticidad focal en niños de 2 años y mayores.
|
Músculos de extremidad superior |
Dosis en unidades/kg/músculo |
Número de inyecciones por músculo |
|
Bíceps Braquial |
0.5 - 2.0 |
2-4 sitios |
|
Braquialis |
0.5 - 2.0 |
1-2 sitios |
|
Braquiorradial |
0.5 - 2.0 |
1-2 sitios |
|
Flexor ulnar del carpo |
0.5 - 2.0 |
1-2 sitios |
|
Flexor radial del carpo |
0.5 - 2.0 |
1-2 sitios |
|
Pronador redondo |
0.5 - 2.0 |
1-2 sitios |
|
Pronador cuadrado |
0.5 - 2.0 |
1-2 sitios |
|
Flexor profundo de los dedos |
0.5 - 2.0 |
1 sitio |
|
Flexor superficial de los dedos |
0.5 - 2.0 |
1 sitio |
|
Flexor largo del pulgar |
0.5 - 2.0 |
1 sitio |
|
Flexor corto del pulgar |
0.5 - 2.0 |
1 sitio |
|
Oponente del pulgar |
0.5 - 2.0 |
1 sitio |
|
Aductor del pulgar |
0.5 - 2.0 |
1 sitio |
|
Aductores de la cadera (aductor largo, aductor corto, aductor magno, isquiotibiales mediales) |
4.0 |
2 sitios |
|
Gastrocnemious Medial Lateral |
2.0 2.0 |
1-2 sitios 1-2 sitios |
Por lo general, la mejoría clínica se presenta dentro de las dos primeras semanas después de la inyección. Se deberán administrar nuevas dosis cuando el efecto clínico de una inyección previa haya disminuido, pero típicamente la frecuencia de inyección no deberá exceder un tratamiento cada tres meses. El grado de espasticidad muscular en el momento de la reinyección puede llegar a hacer necesarias alteraciones de la dosis de BOTOX® y de los músculos a inyectar.
Espasticidad focal de las extremidades superiores asociada con accidente cerebrovascular en adultos: En estudios clínicos controlados y abiertos se utilizaron las siguientes dosis para los músculos individuales (dosis máxima total de 400 U por sesión de tratamiento):
|
Músculo |
Dosis Total; Número de Sitios |
|
Bíceps braquial |
100 – 200 U; 1 a 4 sitios |
|
Flexor profundo de los dedos |
15 – 50 U; 1-2 sitios |
|
Flexor superficial de los dedos |
15 – 50 U; 1-2 sitios |
|
Flexor radial del carpo |
15 – 60 U; 1-2 sitios |
|
Flexor ulnar del carpo |
10 – 50 U; 1-2 sitios |
|
Aductor del pulgar |
20 U; 1-2 sitios |
|
Flexor largo del pulgar |
20 U; 1-2 sitios |
En estudios clínicos controlados y abiertos y en estudios no controlados se administraron dosis que usualmente variaron entre 200 y 240 U en los músculos flexores y de la muñeca (las cuales fueron divididas entre los músculos seleccionados) en una sesión de tratamiento dada.
En estudios clínicos controlados, la mejoría del tono muscular se presentó dentro de las primeras dos semanas, observándose por lo general el efecto pico dentro de un periodo de 4 a 6 semanas. En un estudio de continuación abierto no controlado, la mayoría de los pacientes recibió una nueva inyección después de un intervalo de 12 a 16 semanas, cuando el efecto sobre el tono muscular había disminuido. Los pacientes en cuestión recibieron hasta cuatro inyecciones (con una dosis acumulativa máxima de 960 U) a lo largo de 54 semanas. Si el médico tratante lo considera apropiado, es posible administrar nuevas dosis cuando el efecto de una inyección previa haya disminuido. Por lo general, las nuevas inyecciones no deberán ser administradas antes de 12 semanas. El grado y el patrón de espasticidad muscular en el momento de la reinyección pueden llegar a hacer necesarias alteraciones de la dosis de BOTOX® y de los músculos a inyectar. Se deberá utilizar la dosis eficaz más baja.
Espasticidad Focal de las extremidades inferiores (tobillo) asociada con accidente cerebrovascular en adultos:
La dosis recomendada para el tratamiento de la espasticidad de las extremidades inferiores que involucra el tobillo en adultos es de 300 U, distribuidas en 3 músculos (ver tabla y figura a continuación).
Si el médico tratante lo considera apropiado, se puede repetir el tratamiento con BOTOX® cuando el efecto de una inyección previa haya disminuido, pero no antes de que hayan transcurrido 12 semanas desde la administración de la inyección anterior.
|
Tabla: Dosificación de BOTOX® por músculo para espasticidad de las extremidades inferiores en adultos |
|
|
Músculo |
Dosis recomendada Dosificación total; Número de sitios |
|
Gastrocnemio Cabeza medial Cabeza lateral |
75 Unidades; 3 sitios 75 Unidades; 3 sitios |
|
Soleo |
75 Unidades; 3 sitios |
|
Tibial Posterior |
75 Unidades; 3 sitios |
Figura: Sitios de inyección para espasticidad de las extremidades inferiores en adultos
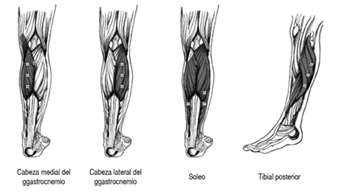
Disfonía Espasmódica: A menos que la inyección sea realizada bajo visualización directa, se utiliza una aguja electromiográfica recubierta con Teflón y la inyección es realizada empleando orientación electromiográfica. Para la disfonía espasmódica aductora, la dosis inicial recomendada es de 1.0 a 2.5 U en un volumen de 0.1 mL inyectado en cada músculo tiroaritenoideo. En los tratamientos subsecuentes, la dosis puede ser ajustada alterando la concentración de acuerdo a las características del paciente y de la respuesta a la terapia previa. Puede que en ocasiones un paciente necesite hasta 3 U por cuerda vocal. Sin embargo, con el paso de los años de tratamiento, muchos pacientes han reducido su dosis hasta una dosis tan baja como 0.2 U por músculo tiroaritenoideo.
Para el tratamiento de la disfonía espasmódica abductora se suelen inyectar 2.0 a 5.0 U de BOTOX® unilateralmente en un músculo cricoaritenoideo posterior a través de un abordaje transcricoideo, supracricoideo o retrocricoideo lateral.
La inyección suele ser administrada con el paciente en posición supina y con una almohada pequeña colocada bajo los hombros para mejorar la exposición laríngea. Para la disfonía espasmódica aductora se identifican los puntos de referencia de la superficie laríngea, incluyendo el cartílago tiroides y cricoides, y en particular el pequeño hueco de la membrana cricotiroidea. La identificación precisa de los puntos de referencia es una parte crítica de este procedimiento y puede llegar a resultar difícil en los individuos con cuello grueso.
También para el tratamiento de la disfonía espasmódica aductora, la aguja de registro EMG es avanzada en la línea media a través de la membrana cricotiroidea, dirigiendo la aguja en dirección rostral y con un ángulo aproximado de 30º en dirección lateral hacia el músculo tiroaritenoideo designado. Para un procedimiento bilateral, la aguja es redirigida hacia el músculo contralateral correspondiente. Una vez dentro del músculo, la actividad electromiográfica de inserción es audible y la colocación puede ser confirmada pidiendo al paciente que articule una “e”. Una vez confirmada la colocación de la aguja se inyecta la dosis requerida de BOTOX® en un volumen de 0.1 mL (por lo general sin exceder 5 U).
En todos los casos de disfonía espasmódica abductora, se deberá realizar una endoscopía antes de cada tratamiento para evaluar la actividad dinámica de cada cuerda vocal y el tamaño de la vía aérea al nivel de la glotis. Típicamente se elige al músculo cricoaritenoideo posterior (PCA) del lado más activo para la terapia. Se deberá utilizar un abordaje retrocricoideo en el cual la aguja de inyección, la cual contiene entre 2 y 5 U de BOTOX® en un volumen de 0.1 mL, es dirigida hacia el PCA describiendo una curva al nivel del cartílago cricoides para posicionarla detrás de la laringe. La laringe puede ser rotada lateralmente en el lado apropiado para mejorar el acceso. Para confirmar la colocación de la aguja, el paciente inhala con fuerza para activar el PCA, lo cual produce un patrón de interferencia EMG característico. A continuación se efectúa la inyección de BOTOX®. Se recomienda realizar exclusivamente inyecciones unilaterales en cada sesión de tratamiento. La determinación del PCA que debe ser inyectado en una sesión de tratamiento dada es realizada a través de una revisión endoscópica previa. Las sesiones de tratamiento son llevadas a cabo exclusivamente cuando la cuerda no inyectada presenta suficiente movimiento para prevenir estridor en el caso de que la cuerda inyectada se vuelva inmóvil. Ocasionalmente, un paciente con disfonía espasmódica abductora presentará un aumento de la actividad del músculo cricotiroideo —la cual también puede ser evaluada por EMG— y posiblemente se beneficiará de inyecciones suplementarias en dicho músculo.
Por lo general, el efecto pico es observado dentro de los 7 días posteriores a una inyección.
Hiperhidrosis:
Hiperhidrosis axilar primaria: La dosis inicial recomendada es de 50 U de BOTOX® es inyectada intradérmicamente utilizando una aguja calibre 30 en alícuotas de 0.1 a 0.2 mL distribuidas uniformemente en múltiples sitios (10-15) con una separación aproximada de 1-2 cm entre sí dentro del área hiperhidrótica de cada axila. El área hiperhidrótica puede ser definida utilizando técnicas de tinción estándar, por ejemplo, la prueba de yodo-almidón de Minor. BOTOX® es reconstituido con solución salina al 0.9% libre de preservantes (100 U/4 mL). Cada dosis es inyectada a una profundidad aproximada de 2 mm y en un ángulo de 45 grados respecto a la superficie de la piel con el lado del bisel hacia arriba para minimizar la filtración y para asegurar que el líquido inyectado permanezca dentro de la dermis.
Por lo general, la mejoría clínica suele presentarse dentro de la primera semana posterior a la inyección. La duración media de la respuesta después de tratamientos repetidos (hasta 4 tratamientos en pacientes tratados con 50 U de BOTOX®) fue de 6-8 meses.
Es posible administrar una nueva inyección de BOTOX® cuando el efecto clínico de una inyección previa haya disminuido y el médico tratante lo considere necesario. Las inyecciones no deberán ser repetidas con una frecuencia que exceda un tratamiento cada dos meses.
Hiperactividad del Músculo Detrusor de la Vejiga:
Trastornos de la vejiga
Los pacientes no deberán presentar infección en el tracto urinario antes del tratamiento. Deberán administrarse antibióticos profilácticos 1-3 días antes del tratamiento, en el día del tratamiento, y 1-3 días después del tratamiento.
Generalmente se recomienda que los pacientes descontinúen el tratamiento anti-plaquetario al menos tres días antes del procedimiento de inyección. Los pacientes con terapia anti-coagulante deben ser controlados adecuadamente para disminuir el riesgo de sangrado.
Vejiga Hiperactiva: Debe realizarse una instilación intravesical de anestésico local diluido con o sin sedación antes de la inyección, de conformidad con la práctica local. Si se realiza una instilación local de anestésico, la vejiga debe ser drenada e irrigada con solución salina estéril antes de la inyección.
La dosis recomendada es 100 Unidades de BOTOX®. La dilución recomendada es 100 Unidades/10 mL con solución salina no preservada 0.9% (ver tabla de dilución). Elimine cualquier sobrante de solución salina.
El BOTOX® reconstituído (100 Unidades/10 mL) es inyectado en el músculo detrusor por medio de un citoscopio flexible o rígido, evitando el trígono. La vejiga debe ser instilada con suficiente solución salina para lograr una adecuada visualización para las inyecciones, pero debe evitarse la sobredistensión.
La aguja de inyección deberá llenarse con aproximadamente 1 mL de BOTOX® reconstituído antes de iniciar las inyecciones (dependiendo de la longitud de la aguja) para remover el aire atrapado.
La aguja deberá ser insertada aproximadamente 2 mm en el detrusor y se deberán realizar 20 inyecciones de 0.5 mL cada una (para un volumen total de 10 mL) con una separación aproximada de 1 cm entre sí (vea la figura). Para la inyección final, se deberá inyectar aproximadamente 1 mL de solución salina normal estéril para administrar la dosis completa. Después de la administración de las inyecciones, la solución salina utilizada para la visualización de la pared de la vejiga no deberá ser drenada para que los pacientes puedan demostrar su capacidad de evacuar antes de abandonar la clínica. El paciente deberá ser observado durante 30 minutos como mínimo después de las inyecciones y hasta que haya ocurrido una evacuación espontánea.
La mejoría clínica puede presentarse dentro de un periodo de 2 semanas. Los pacientes deberán ser considerados para una nueva administración cuando el efecto clínico de las inyecciones previas haya disminuido (la duración media, en estudios clínicos de Fase 3, fue de 166 días [~24 semanas]), pero no antes de 3 meses después de la administración anterior, en la vejiga.
Hiperactividad Neurogénica del Detrusor: Es posible utilizar una instilación intravesical de un anestésico local diluido con o sin sedación, o anestesia general antes de la inyección de conformidad con la práctica local. Si se lleva a cabo una instilación de un anestésico local, la vejiga deberá ser drenada e irrigada con solución salina estéril antes de la inyección.
La dosis recomendada es de 200 U de BOTOX®.
Reconstituya un vial de 200 U de BOTOX® con 6 mL de solución salina al 0.9% libre de preservantes y mezcle suavemente el contenido del vial. Extraiga 2 mL del vial a cada una de tres jeringas de 10 mL. Complete la reconstitución añadiendo 8 mL de solución salina al 0.9% libre de preservantes a cada una de las jeringas de 10 mL y mezcle con cuidado. Siguiendo el procedimiento anterior se obtendrán tres jeringas de 10 mL que contendrán 10 mL (~67 U) cada una, para un total de 200 U de BOTOX® reconstituido. Utilice el producto inmediatamente después de su reconstitución en la jeringa. Deseche toda solución salina no utilizada.
BOTOX® reconstituido (200 U/30 mL) es inyectado en el músculo detrusor por medio de un cistoscopio flexible o rígido evitando el trígono. La vejiga deberá ser instilada con suficiente solución salina para obtener una visualización adecuada para las inyecciones, pero se deberá evitar una distensión excesiva.
La aguja de inyección deberá ser llenada con aproximadamente 1 mL, antes del inicio de las inyecciones (dependiendo de la longitud de la aguja) para eliminar todo aire presente. La aguja deberá ser insertada aproximadamente 2 mm en el detrusor y se deberán realizar 30 inyecciones de 1 mL cada una (para un volumen total de 30 mL) con una separación aproximada de 1 cm entre sí (ver la figura). Para la inyección final, se deberá inyectar aproximadamente 1 mL de solución salina normal estéril para administrar la dosis completa. Después de la administración de las inyecciones, la solución salina utilizada para la visualización de la pared de la vejiga deberá ser drenada. El paciente deberá ser observado durante 30 minutos como mínimo después de las inyecciones.
La mejoría clínica puede presentarse dentro de un periodo de 2 semanas. Los pacientes deberán ser considerados para una nueva administración cuando el efecto clínico de las inyecciones previas haya disminuido (la duración media, en estudios clínicos de Fase 3, fue de 256-295 días ó 36-42 semanas para BOTOX® 200 U), pero no antes de 3 meses después de la administración anterior, en la vejiga.
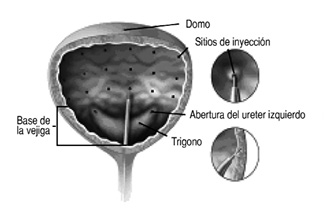
Alternativo en la Profilaxis del Dolor de Cabeza en Migraña Crónica: La dilución recomendada es de 100 unidades/2 mL, con una concentración final de 5 unidades por 0.1 mL. La dosis recomendada para tratar la migraña crónica es de 155 a 195 unidades administradas intramuscularmente (IM) utilizando una aguja calibre 30 estéril de 0.5 pulgadas en inyecciones de 0.1 mL (5 unidades) por sitio. Las inyecciones deberán ser divididas entre 7 áreas específicas de los músculos de la cabeza/cuello según se especifica en la siguiente tabla. Puede ser necesario utilizar una aguja de 1 pulgada en la región del cuello en el caso de los pacientes con músculos del cuello gruesos. Con excepción del músculo procerus, el cual deberá ser inyectado en un sitio (línea media), todos los músculos deberán ser inyectados bilateralmente utilizando la dosis mínima por músculo señalada en la siguiente tabla, ubicándose la mitad del número de sitios de inyección en el lado derecho y la otra mitad en el lado izquierdo de la cabeza y el cuello. Se recomienda repetir el tratamiento cada 12 semanas. De haber una ubicación (o ubicaciones) del dolor predominante(s), es posible administrar inyecciones adicionales en uno o ambos lados en un máximo de 3 grupos de músculos específicos (occipital, temporal y trapecio) hasta alcanzar la dosis máxima por músculo indicada en la siguiente tabla.
Sitios recomendados de inyección para la migraña crónica:

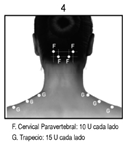
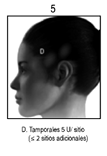
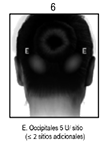
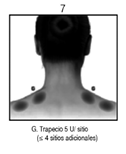
|
Dosis de BOTOX® por músculo para la migrañacrónica |
|
|
Dosis recomendada |
|
|
Área de la Cabeza/Cuello |
Número total de unidades (número de sitios de inyección IMa) |
|
Corrugadorb |
10 unidades (2 sitios) |
|
Prócerb |
5 unidades (1 sitio) |
|
Frontalb |
20 unidades (4 sitios) |
|
Temporalb |
40 unidades (8 sitios); máximo: 50 unidades (hasta 10 sitios) |
|
Occipitalb |
30 unidades (6 sitios); máximo: 40 unidades (hasta 8 sitios) |
|
Grupo de Músculos para espinales cervicalesb |
20 unidades (4 sitios) |
|
Trapeciob |
30 unidades (6 sitios); máximo: 50 unidades (hasta 10 sitios) |
|
Rango de dosis total |
155 unidades a 195 sitios |
|
a Cada sitio de inyección IM = 0.1 mL = 5 unidades (4 sitios) de BOTOX® b Dosis distribuidad bilateralmente en el caso de la dosis mínima |
|
Líneas faciales hiperfuncionales:
Líneas glabelares: BOTOX® reconstituido (50 U/1.25 mL ó 100 U/2.5 mL) es inyectado utilizando una aguja calibre 30 estéril. Se administra un volumen de 0.1 mL (4 U) en cada uno de los cinco sitios de inyección (ver figura): dos inyecciones en cada músculo corrugador y una inyección en el músculo procerus para una dosis total de 20 U.
A fin de reducir la incidencia de ptosis, evítense las inyecciones cerca del músculo elevador palpebral superior, particularmente en los pacientes con complejos depresores del entrecejo de mayor tamaño. Las inyecciones en la parte medial del corrugador y en la parte central de la ceja deberán ser aplicadas al menos 1 cm por encima del reborde óseo supraorbitario.
La mejoría de las líneas verticales entre las cejas (líneas glabelares) suele comenzar dentro de un periodo de 1 a 2 días, incrementándose en intensidad durante la primera semana posterior al tratamiento. La duración del efecto es de aproximadamente 3-4 meses en la mayoría de los pacientes. En algunos pacientes se ha reportado una duración del efecto de hasta 6 meses. La frecuencia de tratamiento no deberá exceder un tratamiento cada tres meses.
Líneas en la frente: BOTOX® reconstituido (50 U/1.25 mL ó 100 U/2.5 mL) es inyectado utilizando una aguja calibre 30 estéril. Por lo general se inyectan 2-6 U intramuscularmente en cada uno de los 4 sitios de inyección en el músculo frontal (cada 1-2 cm a lo largo de cualquiera de los lados de un pliegue profundo de la piel de la frente, 2-3 cm por encima de la ceja) para una dosis total de hasta 24 U.
Líneas Laterales del canto (patas de gallo): BOTOX® reconstituido (50 U/1.25 mL ó 100 U/2.5 mL) es inyectado utilizando una aguja calibre 30 estéril. Por lo general se deben inyectar 2-6 U bilateralmente en cada uno de los sitios de inyección (1-3 sitios) a una profundidad de 2-3 mm lateralmente con respecto al reborde orbitario lateral, donde se observa la mayoría de las líneas de una sonrisa forzada. Las inyecciones deben ser aplicadas al menos 1 cm fuera de la órbita ósea y no deben ser aplicadas en la parte medial de la línea vertical que atraviesa al canto lateral, ni tampoco cerca del margen inferior del cigoma.
Todas las indicaciones
En ausencia del efecto deseado después de la primera sesión de tratamiento (es decir, ausencia de mejoría clínica significativa con respecto a la línea basal antes de que transcurra un mes después de la inyección), se deberán tener en consideración las siguientes acciones:
- Verificación clínica del efecto de la toxina sobre el músculo (o músculos) inyectado(s), lo cual puede incluir un examen electromiográfico por parte de un especialista experimentado en electromiografía.
- Análisis de las potenciales causas de la falta de efecto, por ejemplo, selección inapropiada de los músculos a inyectar, dosis insuficiente, técnica de inyección deficiente, contractura fija, debilidad relativa de los músculos antagonistas y/o formación de anticuerpos neutralizantes contra la toxina;
- Reevaluación de la idoneidad del tratamiento con toxina botulínica tipo A.
Para la segunda sesión de tratamiento, en ausencia de efectos no deseados después de la primera
sesión de tratamiento, el médico deberá tener en consideración lo siguiente:
- Ajuste de la dosis tomando en cuenta el análisis de la falla del tratamiento previo;
- Uso de orientación EMG según sea apropiado;
- Mantenimiento de un intervalo de tres meses entre las dos sesiones de tratamiento.
En caso de falla en el tratamiento o una disminución del efecto después de un nuevo tratamiento, tomando en cuenta los ajustes de la dosis y los objetivos de las inyecciones, se deberán tener en consideración métodos de tratamiento alternativos.
SOBREDOSIS: La sobredosis de BOTOX® es un término relativo y depende de la dosis, del sitio de inyección y de las propiedades de los tejidos subyacentes. Es probable que los signos y síntomas de la sobredosis no sean evidentes inmediatamente después de la inyección. Puede que las dosis excesivas produzcan parálisis neuromuscular local (o distante) generalizada y profunda.
De tener lugar una inyección o ingestión oral accidental, o bien, de sospecharse sobredosis, el paciente deberá ser monitoreado médicamente durante varias semanas para detectar posibles signos o síntomas progresivos de debilidad muscular sistémica, la cual puede ser local o distante del sitio de inyección e incluir ptosis, diplopía, disfagia, disartria, debilidad generalizada o insuficiencia respiratoria. En el caso de estos pacientes se deberá tener en consideración una evaluación médica más a fondo y se deberá instituir una terapia médica apropiada (la cual puede incluir hospitalización) de manera inmediata.
Si la musculatura orofaringea y del esófago se ve afectada, puede que se presente aspiración, lo cual puede conllevar a neumonía por aspiración. Si los músculos respiratorios se paralizan o se debilitan en forma suficiente, puede ser necesario recurrir a intubación y respiración asistida hasta lograr la recuperación. Las medidas de apoyo pueden incluir traqueotomía y/o ventilación mecánica prolongada en adición a otras medidas paliativas generales. Ver también Posología y Método de Administración
INFORMACIÓN ESPECIAL
Incompatibilidades: En ausencia de estudios de incompatibilidad, este medicamento no deberá ser mezclado con otros medicamentos:
Vida Útil y Condiciones de almacenamiento: Los viales sin abrir, deben ser almacenados en refrigeración entre, 2 ºC a 8ºC, ó en congelación entre -5 ºC a - 20ºC. El tiempo de vida útil bajo estas condiciones de almacenamiento es: 3 años
Los viales reconstituidos deben ser almacenados en refrigeración, entre 2°C y 8ºC, y deben utilizarse 72 horas posterior a la reconstitución.
Naturaleza y composición de envase: Vial de vidrio incoloro tipo I de 5ml (50 U)/ 10 ml (200 U) capacidad nominal. Con tapón libre de látex y agrafe de aluminio.
Advertencia: Manténgase fuera del alcance de los niños.
PRESENTACIONES: BOTOX® se suministra en un frasco de uso único. Cada vial contiene complejo de neurotoxina Clostridium botulinum tipo A, secado al vacío en los siguientes tamaños: 50 U, 100 U y 200 U.
Los viales de BOTOX® tienen una película holográfica en la etiqueta que contiene el nombre Allergan dentro de líneas horizontales de color arcoíris. Para poder ver el holograma, rótese el frasco hacia atrás y hacia delante entre los dedos bajo una lámpara de escritorio o una fuente de luz fluorescente.
Nota: En la etiqueta, en el área de la fecha/lote, no hay película holográfica
BOTOX® 100 U:
Registro Sanitario: INVIMA 2014M-014172 R2.
Vigencia: JUN 04-2013
CUM: 45122-01
Código ATC: M03AX01. Agentes relajantes musculares de acción periférica.
Modalidad de Registro: Importar y Vender
BOTOX® BTX-A® 50U:
Registro Sanitario: INVIMA 2009M-0009951
Vigencia: SEP 09-2019
CUM: 20004997-01
Código ATC: M03AX01. Agentes relajantes musculares de acción periférica.
Modalidad de Registro: Importar y Vender
BOTOX® 200U:
Registro Sanitario: INVIMA 2010M-0011586
Vigencia: DEC 01-2015
CUM: 20019432
Código ATC M03AX01. Agentes relajantes musculares de acción periférica.
Fabricado por:
ALLERGAN PHARMACEUTICALS IRELAND
Westport Co., Mayo, Irlanda
Versión: B20050CDS17JUL2014
Fecha de Versión: Febrero de 2015
VIDA ÚTIL Y CONDICIONES DE ALMACENAMIENTO:
Los viales sin abrir, deben ser almacenados en refrigeración entre, 2 ºC a 8ºC, ó en congelación entre -5 ºC a - 20ºC. El tiempo de vida útil bajo estas condiciones de almacenamiento es 3 años.
Los viales reconstituidos deben ser almacenados en refrigeración, entre 2°C y 8ºC, y deben utilizarse 72 horas posterior a la reconstitución.


