

ELIGARD 22.5 MG Y 45 MG
LEUPROLIDA
Polvo para reconstituir inyectable
1 Caja, 2 Sobre(s), 22.5 mg
1 Caja, 2 Sobre(s), 45 mg
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN:
Principio activo: Leuprolide acetato.
Concentración: 22,5 mg y 45 mg.
Forma farmacéutica: Polvo estéril para reconstruir a suspensión inyectable.
Presentación comercial: Caja por 1 sobre con 2 sobres A y B.
Categoría terapéutica: Agente antineoplásico inhibidor de la secreción de gonadotropina.
INDICACIONES: Coadyuvante en el tratamiento de cáncer de próstata avanzado sin tratamiento quirúrgico. Tratamiento del cáncer de seno en mujeres pre y perimenopáusicas que requieren terapia hormonal (supresión ovárica). Pubertad precoz central. Tratamiento de cáncer de próstata siempre que sea necesaria la reducción de valores de testosterona a niveles de castración. Tratamiento de mioma, endometriosis o leimiomatosis uterina.
MECANISMO DE ACCIÓN / FARMACODINAMIA: Acetato de leuprolide es un nonapéptido sintético agonista análogo de la hormona liberadora de gonadotropina (GnRH o LH-RH) que se encuentra en forma natural. Cuando se administra en forma continua inhibe la secreción de gonadotropina hipofisaria y suprime la esteroidogénesis testicular y ovárica.El análogo posee mayor potencia que la hormona natural.
En humanos, la administración de acetato de leuprolide resulta en un incremento inicial de los niveles de circulación de la Hormona Luteinizante (LH) y la hormona folículo-estimulante (FSH), lo cual lleva a un aumento pasajero de los niveles de los esteroides gonadales (testosterona y dilhidrotestosterona en hombres, y estrona y estradiol en mujeres premenopáusicas). No obstante, la administración continua de acetato de leuprolide resulta en la disminución de los niveles de LH y FSH. En los hombres, la testosterona se reduce por debajo del umbral de castración (≤ 50 ng/dL). Estas disminuciones se producen dentro de dos o cuatro semanas después del inicio del tratamiento. Estudios a largo plazo demostraron que la continuación de la terapia con acetato de leuprolide mantiene la testosterona por debajo del nivel de castración por hasta siete años.
ELIGARD® 22.5 mg
En la figura 2, se muestra la farmacocinética/farmacodinamia observada durante dos inyecciones administradas cada tres meses (ELIGARD® 22,5 mg) a 22 pacientes con cáncer de próstata avanzado. La media de las concentraciones séricas de leuprolide aumentó a 127 ng/mL y 107 ng/mL aproximadamente 5 horas después de la aplicación de la primera y la segunda inyección, respectivamente. Después del incremento inicial tras cada inyección, las concentraciones séricas se mantuvieron relativamente constantes (0.2 - 2.0 ng/mL).
ELIGARD® 45 mg
En la figura 3, se muestra la farmacocinética/farmacodinamia observada durante inyecciones administradas al inicio y a los seis meses (ELIGARD® 45 mg) a 27 pacientes con cáncer de próstata avanzado. La media de las concentraciones séricas de leuprolide aumentó a 82 ng/mL y 102 ng/mL (Cmáx) aproximadamente 4,5 horas después de la aplicación de la primera y la segunda inyección respectivamente. Después del incremento inicial tras cada inyección, la media de las concentraciones séricas se mantuvo relativamente constante (0,2 - 2,0 ng/mL)
No hubo evidencia de acumulación significativa durante la repetición de la dosis. Ocasionalmente, se observaron concentraciones plasmáticas no detectables de leuprolide durante la administración de ELIGARD®, pero los niveles de testosterona se mantuvieron en los niveles de castración.
CONTRAINDICACIONES: Hipersensibilidad a la GnRH, a análogos agonistas de GnRH o a cualquier componente de ELIGARD®. Se han informado reacciones anafilácticas a la GnRH sintética o a análogos agonistas de la GnRH.
Contraindicado en mujeres en embarazo o que podrían quedar embarazadas pues puede ocasionar pérdida de embarazo y daño fetal. Sangrado vaginal no diagnosticado, lactancia y menores de 2 años.
Acetato de leuprolide no es un anticonceptivo.
PRINCIPALES REACCIONES ADVERSAS: Se evaluó la seguridad de todas las formulaciones de ELIGARD® en ensayos clínicos en los que participaron pacientes con cáncer de próstata avanzado.
Durante los ensayos clínicos, los lugares de inyección se controlaron de cerca. (Ver Tabla eventos adversos informados en el lugar de la inyección).
|
Tabla 7. Eventos adversos informados en el lugar de inyección |
|||
|
7,5 mg |
22,5 mg |
45 mg |
|
|
Número del estudio |
AGL 9904 |
AGL 9909 |
AGL 0205 |
|
Cantidad de pacientes |
120 |
117 |
111 |
|
Tratamiento |
1 inyección por mes hasta los 6 meses |
1 inyección cada 3 meses hasta los 6 meses |
1 inyección cada 6 meses hasta 12 meses |
|
Cantidad de inyecciones |
716 |
230 |
217 |
|
Quemazón/dolor intenso transitorios |
248 (34,6%) inyecciones; 84% de los casos informados como leves |
50 (21,7%) inyecciones; 86% informados como leves |
35 (16%) inyecciones; 91,4% informados como leves |
|
Dolor (generalmente por poco tiempo y leve) |
4.3% de inyecciones (18,3% de pacientes) |
3,5% de inyecciones (6,0% de pacientes) |
4,6% de inyecciones2 |
|
Eritema (generalmente por poco tiempo y leve) |
2,6% de inyecciones (12,5% de pacientes) |
0,9% de inyecciones (1,7% de pacientes)1 |
|
|
Hematoma (leve) |
2,5% de inyecciones (11,7% de pacientes) |
1,7% de inyecciones (3,4% de pacientes) |
2,3% de inyecciones3 |
|
Prurito |
1,4% de inyecciones (9,2% de pacientes) |
0,4% de inyecciones (0,9% de pacientes) |
|
|
Induración |
0,4% de inyecciones (2,5% de pacientes) |
||
|
Ulceración |
0,1% de inyecciones (> 0,8% de pacientes) |
||
|
1. Se informó eritema tras 2 inyecciones de ELIGARD® 22,5 mg. Un informe caracterizó el edema como leve y se resolvió dentro de 7 días. El otro informe caracterizó el eritema como moderado y se resolvió dentro de 15 días. Ningún paciente experimento eritema en inyecciones múltiples. 2. El dolor pasajero fue informado como leve en 9 de 10 eventos (90%) y moderado en 1 de 10 (10%) eventos tras la inyección de ELIGARD® 45 mg. 3. Se informan hematomas leves tras 5 inyecciones (2,3%) y hematomas moderadas tras 2 inyecciones (<1%) de ELIGARD® 45 mg. |
|||
Estos eventos adversos no fueron recurrrentes con el transcurso del tiempo. Ningún paciente discontinuó la terapia debido a un evento adverso en el lugar de la inyección.
Los siguientes eventos adversos sistémicos posible o probablemente relacionados se produjeron durante los ensayos clínicos con ELIGARD® y se informaron en > 2% de los pacientes (Tabla 8). A menudo, es difícil evaluar la casualidad en pacientes con cáncer de próstata metastásico. Se excluyen las reacciones que no se consideraron relacionadas con el medicamento.
|
Tabla 8. Resumen de eventos adversos sistémicos posible o probablemente relacionados, informados por >2% de pacientes tratados con ELIGARD® |
|||||
|
7,5 mg |
7,5 mg |
22,5 mg |
45 mg |
||
|
Número del estudio |
AGL 9904 |
AGL 9802 |
AGL 9909 |
AGL 0205 |
|
|
Cantidad de pacientes |
120 |
8 |
117 |
111 |
|
|
Tratamiento |
1 Inyección por mes hasta 6 meses |
1 inyección (pacientes castrados quirúrg.) |
1 inyección cada 3 meses hasta 6 meses |
1 inyección cada 6 meses hsata 12 meses |
|
|
Sistema orgánico |
Evento adverso |
Número de pacientes (porcentaje) |
|||
|
Sistemico |
Malestar y fatiga |
21 (17,5%) |
7 (6,0%) |
13 (11,7%) |
|
|
Debilidad |
4 (3.6%) |
||||
|
Sistema nervioso |
Mareos |
4 (3.3%) |
|||
|
Vascular |
Sofocos/sudor |
68 (56,7%)* |
2 (25,0%)* |
66 (56,4%)* |
64 (57,7%)* |
|
Renales/Urin. |
Frecuencia urinaria |
3 (2,6%) |
|||
|
Gastrointestinal |
Náuseas |
4 (3,4%) |
|||
|
Gastroenteritis/colitis |
3 (2,5%) |
||||
|
De la piel |
Prurito |
3 (2,6%) |
|||
|
Piel fría |
- |
- |
- |
- |
|
|
Sudores nocturnos |
3 (2,7%) |
||||
|
Alopecia |
- |
- |
- |
- |
|
|
Musculoesquelético |
Astralgia |
4 (3,4%) |
|||
|
Mialgia |
5 (4,5%) |
||||
|
Dolor en las extremidades |
3 (2,7%) |
||||
|
Del aparato reproductor |
Atrofia testicular |
6 (5,0%) |
8 (7,2%)* |
||
|
Ginecomastia |
4 (3,6%)* |
||||
|
* Consecuencias esperadas con la suspención de testosterona. En las poblaciones de pacientes estudiantes que recibieron ELIGARD® 7,5 mg, se informaron 86 eventos adversos de sofocos/sudor en 70 pacientes. De estos, 71 eventos (83%) fueron leves; 14 (16%) fueron moderados; 1 (1%) fue severo. En la población de pacientes estudiada que recibio ELIGARD® 22,5 mg, se informaron 84 eventos adversos de sofocos/sudor en 66 pacientes. De estos, 73 eventos (87%) fueron leves; 11 (13%) fueron moderados; ninguno fue severo. En la población de pacientes estudiada que recibió ELIGAARD® 45 mg, se informaron 89 eventos adversos de sofocos en 64 pacientes. Des estos, 62 eventos (70%) fueron leves. |
|||||
Además los siguientes eventos adversos sistémicos posible o probablemente relacionados fueron informados en <2% de los pacientes tratados con ELIGARD® durante los estudios clínicos:
|
Sistema y órganos |
Evento adverso |
|
Generales |
Transpiración, insomnio, síncope, escalofrío, debilidad, letargia. |
|
Gastrointestinales |
Flatulencia, constipación, despepsia. |
|
Hematológicos |
Disminución en el recuento de globulos rojos, hematocritos y hemoglobina. |
|
Matabólicos |
Aumento de peso. |
|
Musculoesqueléticos |
Temblores, dolor de esplada, dolor en las articulaciones, atrofia muscular, dolor en los miembros. |
|
Nerviosos |
Alteración del olfato y el gusto, depresión, vértigo. |
|
Psiquiátricos |
Insomnio, depresión, pérdida de la libido* |
|
Renales/urinarios |
Dificualtad al orinar, dolor al orinar, orina escasa, espasmo vesical, sangre en orina, retención urinaria, micción urgente, incontinencia, nocturia, nocturia agravada. |
|
Del aparato reproductor/urogenitales |
Dolor/sensibilidad en los testículos, impotencia*, disminución de la libido*, ginecomastia*, dolor/sensibilidad en las mamas*, atrofia testicular*, disfunción eréctil, trastornos en el pene*, tamaño reducido del pene. |
|
De la piel |
Alopecia, piel fría, sudores nocturnos*, aumento de la transpiración*. |
|
Vasculares |
Hipertensión, hipotensión. |
|
* Consecuencias farmacológicas esperadas de la supresión de testosterona. |
|
Cambios en la densidad ósea: Se informó una disminución de la densidad ósea en la literatura médica en hombres sometidos a una orquiectomía o que fueron tratados con un análogo agonista de la GnRH. Puede preverse que periodos prolongados de castración médica en hombres tengan efectos sobre la densidad ósea.
Experiencia poscomercialización: Durante la vigilancia posterior a la comercialización, se informaron casos raros de apoplejía hipofisiaria (un síndrome clínico derivado del infarto de la hipófisis) después de la administración de agonistas de la hormona liberadora de gonadotropina. En la mayoría de estos casos, se diagnosticó un adenoma hipofisiario. La mayoría de los casos de apoplejía hipofisiaria se produjeron dentro de las 2 semanas de la primera dosis y algunos dentro de la primera hora. En estos casos, la apoplejía hipofisiaria se presentó como dolor de cabeza repentino, vómitos, cambios visuales, oftalmoplejía, alteración del estado mental y, a veces, colapso cardiovascular. Se requirió atención médica inmediata.
También se informaron casos de convulsiones en el contexto de la poscomercialización.
INTERACCIONES: No se han llevado a cabo estudios de interacción farmacocinética con ELIGARD®.
PRECAUCIONES Y ADVERTENCIAS: Interacciones entre drogas y prueba de laboratorio: La terapia con acetato de Leuprolide resulta en la supresión del sistema hipofisiario-gonadal. Pueden verse afectados los resultados de estudios diagnósticos de las funciones hipofisarias, gonadotropias y gonadales llevados a cabo durante y después de la terapia con Leuprolide.
ELIGARD®, como otros análogos de LH-RH, provocó un incremento pasajero en las concentraciones séricas de testosterona, hasta del 50% por encima del valor basal, durante la primera y segunda semana de tratamiento. Por lo tanto, los pacientes pueden experimentar exacerbaciones de los signos y síntomas de enfermedades o aparición de nuevos síntomas durante las primeras semanas de tratamiento incluido dolor óseo, neuropatía, hematuria u obstrucción a la salida de la vejiga. En el tratamiento paliativo del cáncer de próstata avanzado, utilizando agonistas de GnRH, se observaron casos de obstrucción ureteral y/o compresión medular, que pueden contribuir a la parálisis con o sin complicaciones fatales. Los pacientes con lesiones vertebrales metastásicas y/o con obstrucción del tracto urinario deben ser observados durante las primeras semanas de terapia e instaurar el tratamiento estándar para estas complicaciones.
Pruebas de laboratorio: Se debe controlar la respuesta a ELIGARD® midiendo las concentraciones séricas de testosterona y del antígeno prostático de forma periódica. En la mayoría de los pacientes, los niveles de testosterona se incrementan por encima de los valores basales durante la primera semana, y disminuyeron de ahí en adelante a niveles basales o por debajo de estos hacia finales de la segunda a tercera semana. Por lo general, los niveles de castración se alcanzan dentro de las dos a cuatro semanas. Los niveles de castración de testesterona usualmente se mantienen durante el tratamiento.
Los resultados de las determinaciones de testosterona dependen del método de valoración. Es aconsejable tener en cuenta el tipo y la precisión del método de valoración para tomar decisiones clínicas y terapéuticas adecuadas.
Hiperglicemia y diabetes: Se han informado casos de hiperglicemia y un mayor riesgo de desarrollar diabetes en hombres que reciben agonistas de GnRH. La hiperglicemia puede representar un desarrollo de diabetes mellitus o un empeoramiento del control de la glicemia en pacientes con diabetes. Se debe monitorear periódicamente la glucosa y/o la hemoglobina glicosilada (HbA1c) y manejar con las prácticas corrientes vigentes para el tratamiento de la hiperglicemia y/o la diabetes.
Enfermedades cardiovasculares: Se ha informado un mayor riesgo de desarrollar infarto del miocardio, muerte súbita cardíaca y accidente cerebro vascular en relación con el uso de agonistas de GnRH en hombres. El riesgo parece bajo en base a los odds ratios informados y debe evaluarse cuidadosamente junto con los factores de riesgo cardiovasculares, al momento de determinar el tratamiento para el cáncer de próstata. Los pacientes deben ser monitoreados para detectar síntomas o signos que puedan indicar desarrollo de enfermedad cardiovascular y deben manejarse de acuerdo a la práctica clínica vigente.
Efecto sobre intervalo QT/QTc: La terapia de deprivación androgénica puede prolongar el intervalo QT/QTc. Se debe evaluar el beneficio de la terapia antiandrogénica vs el riesgo en pacientes con síndrome de QT largo, insuficiencia cardíaca congestiva, anomalías electrolíticas frecuentes y en pacientes que toman medicamentos conocidos por prolongar el intervalo QT (Medicamentos clase IA como quinidina, procainamida o clase III como amiodarona y antiarrítmicos como Sotalol ). Considerar el monitoreo periódico de electrolitos y electrocardiograma.
Densidad Mineral Ósea: Pueden ocurrir cambios en la densidad ósea como en cualquier estado hipoestrogénico en mujeres, y en el uso a largo plazo en el cáncer de próstata en el hombre. No existe evidencia de reversión luego del retiro en hombres. En mujeres puede ser reversible.
Convulsiones: Se han descrito convulsiones en reportes post-comercialización, en hombres y mujeres y en la edad pediátrica, con antecedentes de convulsiones, de accidentes cerebrovasculares, anomalías del sistema nervioso central o tumores y con medicación concomitante como Bupropión o inhibidores de la recaptación de serotonina.
Mujeres con endometriosis o fibromas uterinos: Durante la primera fase del tratamiento los esteroides sexuales aumentan temporalmente por lo que puede presentarse un aumento de los síntomas y signos clínicos, pero estos se atenúan con la terapia continuada. Se han reportado casos de fuerte sangrado genital que pueden requerir tratamiento médico o quirúrgico en casos de leiomioma submucoso.
Reproducción asistida: Se debe hacer la inducción de la ovulación bajo la supervisión de un especialista en esta área. En algunas mujeres con predisposición, principalmente con ovario poliquístico, el tratamiento puede causar una excesiva respuesta folicular. En caso de hiperestimulación del ovario la Gonadotrofina se debe interrumpir por unos pocos días mientras se continúa el tratamiento con acetato de leuprolide para prevenir la elevación de la hormona luteinizante. La respuesta del ovario a la administración de acetato de leuprolide y gonadotropinas puede variar de una mujer a otra y entre ciclos en una misma mujer.
Niños: El incumplimiento del régimen del tratamiento o la dosificación inadecuada pueden producir un control inadecuado del proceso puberal y pueden regresar signos como desarrollo de las mamas y crecimiento testicular. Las consecuencias a largo plazo del control inadecuado de la secreción gonadal se desconocen pero pueden incluir un compromiso posterior de la estatura adulta.
Debe monitorearse la respuesta a acetato de leuprolide 1 a 2 meses después del comienzo del tratamiento con una prueba de estimulación de la GNRH y con niveles de esteroides sexuales. La evaluación de la progresión de la edad ósea debe hacerse cada 6 a 12 meses.
Los esteroides sexuales pueden aumentar por arriba de niveles pre-puberales si la dosis es inadecuada. Una vez establecida la dosis correcta, los niveles de gonadotropina y de esteroides sexuales disminuirán a valores prepuberales.
DOSIS Y VÍA DE APLICACIÓN: ELIGARD® se administra de manera SC y proporciona una liberación continua de acetato de Leuprolide durante un período de tratamiento de tres o seis meses. La inyección distribuye la dosis de acetato de Leuprolide incorporada en una formulación polimérica.
|
Dosis recomendada de ELIGARD® |
||
|
Dosis |
22,5 mg |
45 mg |
|
Dosis Recomendada |
1 inyección cada 3 meses |
1 inyección cada 6 meses |
Una vez mezclado ELIGARD® debe desecharse si no se administra dentro de los 30 minutos.
Al igual que con otros fármacos administrados mediante inyección sub-cutánea, el lugar de la inyección debe variar periódicamente. El lugar elegido para la aplicación debe ser un área con suficiente tejido subcutáneo blando o flojo. (parte superior o media de abdomen. Evitar áreas con tejido subcutáneo fibroso o grueso o lugares que podrían restregarse o comprimirse (ej. cinturón o ropa ajustada a la cintura).
SOBREDOSIFICACIÓN: En ensayos clínicos en los que se utilizaron inyecciones subcutáneas diarias de acetato de leuprolide en pacientes con cáncer de próstata, las dosis que llegan a 20 mg/día por hasta dos años no provocan efectos adversos diferentes de los observados con la dosis de 1 mg/día.
DESCRIPCIÓN: ELIGARD® es una fórmula polimérica estéril de acetato de leuprolide, un agonista de la GnRH, para inyección subcutánea. Está diseñado para liberar acetato de leuprolide a una velocidad controlada a lo largo de un periodo terapéutico de uno, tres, cuatro o seis meses.
El acetato de leuprolide es un análogo nonapéptido sintético de la hormona liberadora de gonadotropina (GnRH) que se encuentra en forma natural. Cuando se administra en forma continua, inhibe la secreción de gonadotropina hipofisiaria y suprime la esteroidogénesis testicular y ovárica. El análogo posee mayor potencia que la hormona natural. El nombre químico es 5-oxo-L-prolil-L-histodil-L-triptofil-L-seril-L-tirosil-D-leucil-L-arginil-N-etil-L-acetato de prolinamida (sal).
ELIGARD® es prellenado y suministrado en dos jeringas estériles distintas cuyo contenido se mezcla inmediatamente antes de la administración. Las dos jeringas se unen y el producto de dosis única se mezcla hasta que esté homogéneo. ELIGARD® se administra por vía subcutánea, donde se formaun sólido depósito para la liberación del fármaco.
Una jeringa contiene el sistema de distribución ATRIGEL® y la otra contiene acetato de leuprolide. ATRIGEL® es un sistema de distribución polimérico (sin contenido de gelatina) que consiste en una formulación polímera poli (DL-láctido-co-glicólido) (PLGH o PLG) biodegrafable disuelta en un solvente biocompatible, N-metil-2-pirrolidona (NMP).
PRESENTACIÓN:
ELIGARD® 22,5 mg Caja por 1 sobre con 2 sobres A y B. (Reg. San. INVIMA 2016M-0004777 R1).
ELIGARD® 45 mg Caja por 1 sobre con 2 sobres A y B. (Reg. San. INVIMA 2018M-0013021 R1).
*** Acta Comisión Revisora de Medicamentos # 23 de 2015 ( 3.2.16)
Elaborado en:
TOLMAR Inc: 701 Centre Avenue, Fort Collins, CO 80526, USA
Distribuido por ADIUM S.A.S
ADIUM S.A.S
Para mayor información por favor comunicarse a
Carrera 16 No. 85 -96 - Bogotá, D.C. - Teléfono: PBX 601-6460505
página web: www.adium.com.co
ALMACENAMIENTO: Conservar entre 2 y 8 °C. Mantener fuera del alcance de los niños.
INSTRUCCIONES PARA ADMINISTRACIÓN: ELIGARD® es prellenado y suministrado en 2 jeringas estériles distintas cuyo contenido se mezcla inmediatamente antes de la administración. Las dos jeringas se unen y el producto de dosis única se mezcla hasta que esté homogéneo.
ELIGARD® se administra por vía subcutánea donde se forma un sólido depósito para la liberación del fármaco:
ELIGARD® 22,5 mg:
- Cada jeringa reconstituida dispensa:
Leuprolida acetato 22,5 mg
50:50 Poli (DL-láctido-co-glicólido) 158,6 mg
N-metil-2-pirrolidona 193,9 mg
- Cada jeringa prellenada A (ATRIGEL® sistema de liberación) contiene:
(75:25) Poli (DL-láctido-co-glicólido) 206 mg
N-metil-2-pirrolidona 251 mg
- Cada jeringa prellenada B contiene:
Leuprolida acetato 29,2 mg
ELIGARD® 22,5 mg se presenta con dos jeringas prellenadas separadas, estériles, cuyos contenidos son mezclados de inmediato previo a la administración. Ambas jeringas son unidas y el producto de dosis única es mezclado hasta homogeneización. Leuprolida acetato 22,5 mg es administrado una vez cada tres meses por vía subcutánea donde forma un depósito sólido de liberación de droga.
La jeringa A contiene el Sistema de Liberación ATRIGEL y la jeringa B contiene leuprolida acetato. ATRIGEL es un sistema de liberación polimérico (sin contenido de gelatina) que consiste en una formulación polimérica (poli-DL-láctido-co-glicólido) (PLG) biodegradable disuelto en un solvente biocompatible, N-metil-2-pirrolidona (NMP). PLG es un co-polímero con una ratio molar 75:25 de DL-láctido conteniendo grupos terminales de carboxilo. La segunda jeringa contiene leuprolida acetato y el producto constituido libera 22,5 mg de leuprolida acetato en el momento de la inyección subcutánea (equivalente a 21 mg de leuprolida base libre) disuelto en 242 mg de N-metil-2-pirrolidona y 198 mg poli (DL-lactido-co-glicólido). El peso aproximado de la formulación administrada es de 375 mg. El volumen aproximado a inyectar es de 0,375 ml.
ELIGARD® 45 mg:
- Cada jeringa reconstituida dispensa:
Leuprolida acetato 45 mg
85:15 Poli (DL-láctido-co-glicólido) 165 mg
N-metil-2-pirrolidona 165 mg
- Cada jeringa prellenada A (ATRIGEL® sistema de liberación) contiene:
(85:25) Poli (DL-láctido-co-glicólido) 217 mg
N-metil-2-pirrolidona 217 mg
- Cada jeringa prellenada B contiene:
Leuprolida acetato 59,2 mg
ELIGARD® 45 mg se presenta con dos jeringas prellenadas separadas, estériles, cuyos contenidos son mezclados de inmediato previo a la administración. Ambas jeringas son unidas y el producto de dosis única es mezclado hasta homogeneización. Leuprolida acetato 45 mg es administrado una vez cada seis meses por vía subcutánea donde forma un depósito sólido de liberación de droga.
La jeringa A contiene el Sistema de Liberación ATRIGEL y la jeringa B contiene leuprolida acetato. ATRIGEL es un sistema de liberación polimérico (sin contenido de gelatina) que consiste en una formulación polimérica (poli-DL-láctido-co-glicólido) (PLG) biodegradable disuelto en un solvente biocompatible, N-metil-2-pirrolidona (NMP). PLG es un co-polímero con una ratio molar 85:15 de DL-láctido conteniendo grupos terminales de carboxilo. La segunda jeringa contiene leuprolida acetato y el producto constituido libera 45 mg de leuprolida acetato en el momento de la inyección subcutánea (equivalente a 42 mg de leuprolida base libre) disuelto en 165 mg de N-metil-2-pirrolidona y 165 mg poli (DL-lactido-co-glicólido). El peso aproximado de la formulación administrada es de 375 mg. El volumen aproximado a inyectar es de 0,375 ml.
Procedimiento de mezclado:
IMPORTANTE: Deje que el producto alcance temperatura ambiente antes de usarlo. Una vez mezclado, el producto debe administrarse dentro de los 30 minutos.
Siga las instrucciones para garantizar la preparación adecuada de
ELIGARD® antes de la administración:
ELIGARD® se envasa en dos bandejas termoformadas. Cada caja contiene:
• Una jeringa estéril (Jeringa A), prellenada con el sistema de distribución ATRIGEL®.
• Una jeringa estéril (Jeringa B) prellenada con polvo de acetato de leuprolide Conserve este prospecto.Es posible que necesite volver a leerlo.
• Un vástago del émbolo blanco largo para uso con la Jeringa B.
• Una aguja estéril o Una aguja estéril de seguridad.
• Paquetes de desecantes.
1. En un lugar limpio, abra todos los envases exteriores y extraiga el contenido. Elimine los paquetes de desecantes.
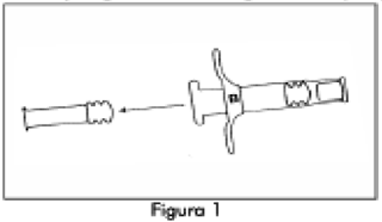
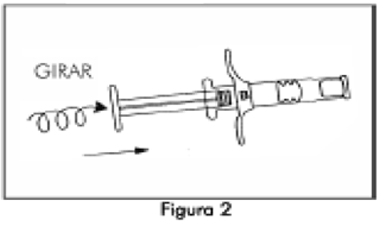
2. Retire el vástago del émbolo corto azul con el tapón gris adherido de la Jeringa B y deseche (Figura 1). Gire el vástago del émbolo largo, blanco, de reemplazo, dentro del tapón primario gris que queda en la Jeringa B (Figura 2).
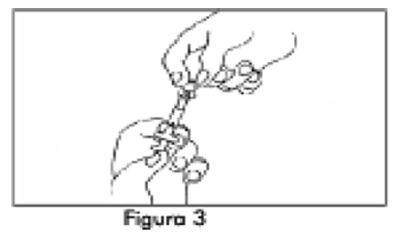
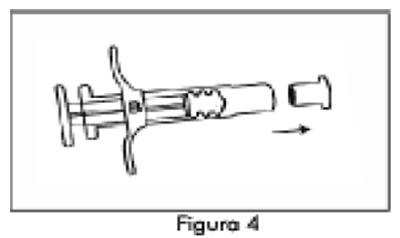
3. Desenrosque el protector transparente de la jeringa A (Figura 3). Elimine el protector gris de goma de la Jeringa B (Figura 4).
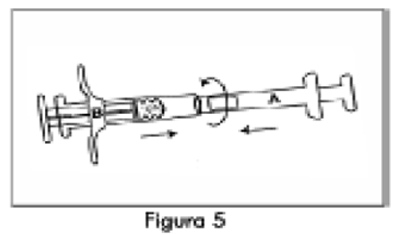
4. Acople bien las dos jeringas presionando y girando hasta que queden aseguradas (Figura 5).
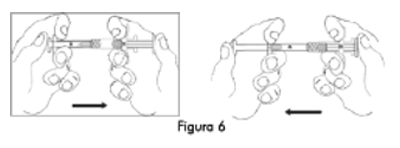
5. Inyecte el contenido de líquido de la Jeringa A en la Jeringa B que contiene acetato de leuprolide. Mezcle bien el producto empujando el contenido hacia atrás y adelante entre las jeringas para obtener una suspensión uniforme (Figura 6). Una vez que se mezcla por completo, la suspensión tendrá un aspecto de color marrón claro a marrón (ELIGARD® 7,5 mg) o incoloro a amarillo claro (ELIGARD® 22,5 mg y 45 mg). Por favor tenga en cuenta que el producto debe mezclarse tal como se describe. No lo agite ya que NO se mezclará adecuadamente.
6. Luego del mezclado, mantenga las jeringas en forma vertical con la jeringa B abajo. Las jeringas deben mantenerse acopladas de manera segura.Vierta todo el producto mezclado en la Jeringa B (jeringa corta, ancha) oprimiendo el émbolo de la jeringa A y retirando suavemente el émbolo de la jeringa B.Desenrosque la Jeringa A mientras continúa empujando el émbolo de la Jeringa A (Figura 7). Nota: En la formulación quedarán pequeñas burbujas de aire.Esto es aceptable.
[Corresponde al kit de ELIGARD® de uso único de un sistema de mezcla de dos jeringas con aguja estéril]

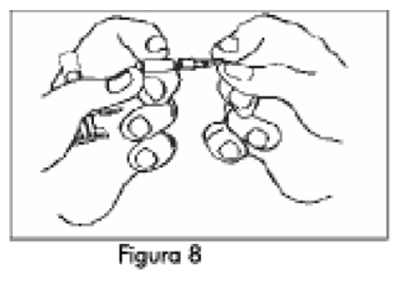
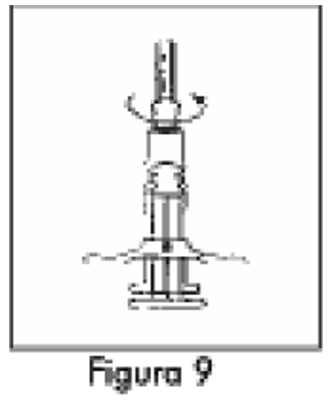
7. Mantenga la Jeringa B en posición vertical. Quite el protector de la base del cartucho de la aguja estéril haciéndolo girar (Figura 8). Coloque el cartucho de la aguja en el extremo de la Jeringa B (Figura 9) empujando y haciendo girar la aguja hasta que esté ubicada firmemente. No haga sobregirar la aguja en la jeringa porque puede salirse la tapa. Retire el protector transparente del cartucho de la aguja antes de la administración (Figura 10).
[Corresponde al kit de ELIGARD de uso único de un sistema de mezcla de dos jeringas con aguja estéril]
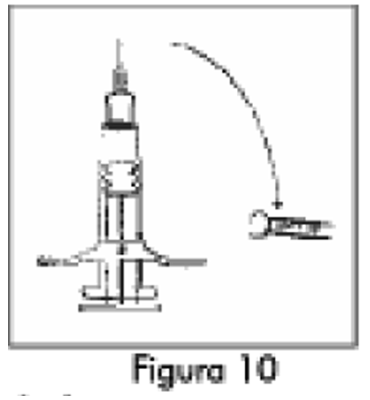
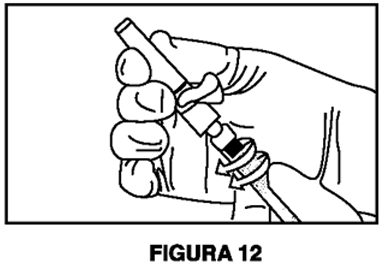
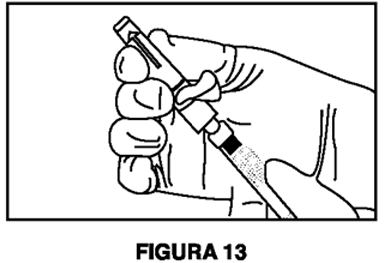
8. Mantenga la jeringa B en posición vertical.Abra el paquete de la aguja de seguridad estéril quitando la solapa de papel y retire la aguja de seguridad (Figura 11).Asegure la aguja al extremo de la jeringa B sosteniendo la funda protectora de la aguja y girando la jeringa en el sentido de las agujas del reloj para colocar completamente la aguja (Figura 12).Retire la funda protectora de la aguja antes de la administración (Figura 13).
Procedimiento de administración:
IMPORTANTE: Deje que el producto alcance temperatura ambiente antes de mezclarlo. Una vez mezclado,el producto debe administrarse dentro de los 30 minutos.
1. Como lugar de aplicación, elija un lugar en el abdomen, parte superior de los glúteos u otro lugar con suficiente tejido subcutáneo que no posea pigmentación, nódulos, lesiones o vello en exceso. Como se puede variar el lugar de inyección para la inyección, elija un área que no se haya utilizado recientemente.
2. Limpie el lugar de inyección con un algodón embebido en alcohol.
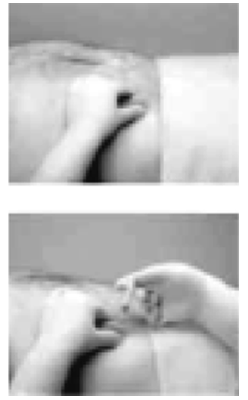
3. Utilizando el pulgar y el índice de su mano no dominante, tome y abulte el área de la piel en torno al lugar de inyección.
4. Utilizando su mano dominante, inserte la aguja rápidamente con un ángulo de 90º en la superficie de la piel. La profundidad de la penetración dependerá del volumen de tejido subcutáneo y de la longitud de la aguja. Una vez insertada la aguja, suelte la piel con su mano no dominante.
5. Inyecte el fármaco empujando lenta y sostenidamente.Presione el émbolo hasta que la jeringa esté vacía.
6. Retire la aguja rápidamente en el mismo ángulo de 90º utilizado para la inserción.
El paso 7 aplica solamente al kit de uso único de ELIGARD® de un sistema de mezcla de dos jeringas con aguja de seguridad estéril.
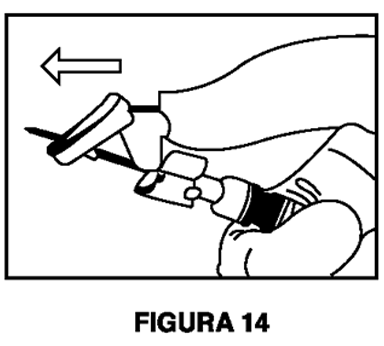
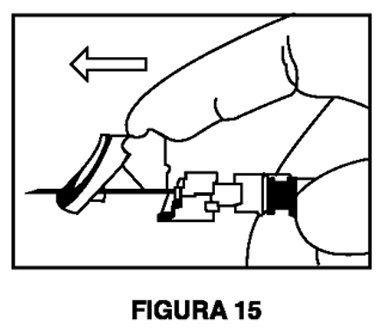
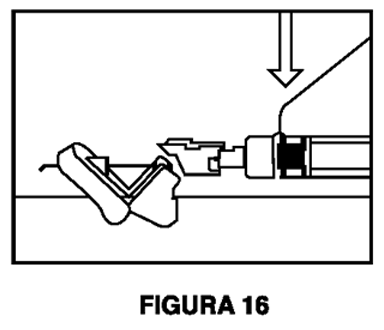
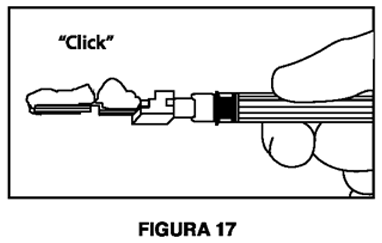
7. Inmediatamente después de retirar la aguja, active la cubierta de seguridad en la aguja usando un pulgar (Figura 14) o dedo (Figura 15) o superficie plana (Figura 16) para empujarla hasta que cubra por completo la punta de la aguja y quede fija en su lugar. El sonido de un clic verifica la posición de bloqueo de la cubierta de seguridad (Figura 17).
El paso 7 aplica solamente al kit de uso único de ELIGARD® de un sistema de mezcla de dos jeringas con aguja de seguridad estéril
8. Deseche todos los componentes de manera segura en un recipiente apropiado para material con riesgo biológico.


