

ENSPRYNG
SATRALIZUMAB
Solución inyectable
Caja, 1 Jeringa prellenada, 120 mg
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA: SOLUCIÓN para inyección subcuatánea que contiene 120 mg de satralizumab
INDICACIONES TERAPÉUTICAS: ENSPRYNG está indicado como monoterapia o en combinación con terapia con corticoides orales para el tratamiento de los trastornos del espectro de la neuromielitis óptica (TENMO) en adultos que son antiacuaporina 4 (AQP4) seropositivos.
USO EN POBLACIONES ESPECIALES: Mujeres y varones con capacidad de procrear: Fertilidad: No se dispone de datos clínicos sobre el efecto de ENSPRYNG en la fertilidad humana. Los estudios en animales no mostraron alteraciones de la fertilidad masculina o femenina (v. Deterioro de la fertilidad).
Embarazo: No hay datos relativos al uso de ENSPRYNG en mujeres embarazadas.
Los estudios en monos indican que no existen efectos perjudiciales con respecto a la toxicidad para la reproducción (v. Toxicidad para la reproducción).
No se recomienda el uso de ENSPRYNG durante el embarazo a menos que el posible beneficio para la madre compense el posible riesgo para el feto.
Lactancia: Se desconoce si ENSPRYNG se excreta en la leche materna humana o si presenta absorción sistémica tras su ingestión. Sin embargo, dado que las IgG se excretan en la leche humana y que existen datos preclínicos de excreción en la leche (v. Toxicidad para la reproducción), deberá tomarse la decisión de interrumpir la lactancia o suspender el tratamiento con ENSPRYNG tras sopesar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre.
Uso en pediatría: No se han estudiado suficientemente la seguridad ni la eficacia de ENSPRYNG en la población de menos de 18 años.
Uso en ancianos: La seguridad y la eficacia de ENSPRYNG se han estudiado en pacientes ancianos de hasta 74 años (v. Instrucciones posológicas especiales y Farmacocinética en poblaciones especiales).
No se han estudiado la seguridad ni la eficacia de ENSPRYNG en pacientes ancianos mayores de 74 años (v. Instrucciones posológicas especiales).
Insuficiencia renal: No se han estudiado formalmente la seguridad ni la eficacia de ENSPRYNG en pacientes con insuficiencia renal, pero dado que ENSPRYNG es un anticuerpo monoclonal y se elimina por catabolismo (y no por excreción renal), no es previsible que sea necesario ajustar la dosis en los pacientes con insuficiencia renal. En los ensayos clínicos se incluyó a pacientes con insuficiencia renal leve; la farmacocinética del satralizumab no resultó afectada en estos pacientes (v. Instrucciones posológicas especiales y Farmacocinética en poblaciones especiales).
Insuficiencia hepática: No se han estudiado la seguridad ni la eficacia de ENSPRYNG en pacientes con insuficiencia hepática (v. Instrucciones posológicas especiales y Farmacocinética en poblaciones especiales).
DATOS FARMACÉUTICOS: Conservación: Condiciones establecidas en cada región.
Conservar a 2-8 °C hasta el momento de su uso.
Los envases de ENSPRYNG sin abrir pueden sacar y volver a guardar en la nevera en caso necesario. Si se conserva a temperatura ambiente, el tiempo total combinado sin refrigeración no debe exceder de 8 días a una temperatura que no supere los 30 °C.
Conservar la JPC en la caja de cartón exterior para protegerla de la luz.
No congelar. No agitar.
Periodo de validez: Condiciones establecidas en cada región.
Este medicamento no debe utilizarse después de la fecha de caducidad (CAD) que figura en el envase.
PROPIEDADES FARMACOCINÉTICAS: La farmacocinética de ENSPRYNG se ha caracterizado en voluntarios sanos japoneses y de raza blanca, y en pacientes con NMO y TENMO. La farmacocinética en pacientes con NMO y TENMO tratados con la dosis recomendada se caracterizó mediante métodos de análisis de farmacocinética poblacional utilizando una base de datos de 104 pacientes.
La evolución de la concentración de ENSPRYNG en función del tiempo en los pacientes con NMO o TENMO se describió con exactitud mediante un modelo bicompartimental de FC poblacional con eliminación paralela lineal y mediada por la diana (Michaelis-Menten) y absorción s. c. de primer orden. Los parámetros de aclaramiento y volumen de ENSPRYNG aumentaron en una relación alométrica en función del peso corporal (mediante la función de potencia con un coeficiente de potencia fijo de 0,75 y 1 para los parámetros de aclaramiento y volumen, respectivamente). Se demostró que el peso corporal era una covariable significativa, observándose un aumento del aclaramiento del 71,3 % y del Vdc del 105 %, respectivamente, en los pacientes que pesaban 123 kg (percentil 97,5 de la distribución del peso) en comparación con un paciente de 60 kg.
La farmacocinética en estado de equilibrio se alcanzó después del periodo de carga (8 semanas) para la Cmín, la Cmáx y el ABC como sigue (media [±DE]): Cmín: 19,7 (12,2) µg/ml, Cmáx: 31,5 (14,9) µg/ml y ABC: 737 (386) µg/ml/día. La inmunoterapia de base no incluyó en la farmacocinética (véase el apartado 2.8 Interacciones con otros medicamentos y otras formas de interacción)
Absorción: La constante de absorción de ENSPRYNG fue de 0,251 día-1 (IC del 95 %: 0,216-0,285), lo que equivale a una semivida de absorción de unos 3 días con la dosis recomendada (véase el apartado 2.2 Posología y administración). La biodisponibilidad fue elevada (85,4 %, IC del 95 %: 0,795-0,953).
Distribución: ENSPRYNG sigue una distribución bifásica. El volumen de distribución central fue de 3,46 l (IC del 95 %: 3,21-3,97) y el volumen de distribución periférico, de 2,07 l (IC del 95 %: 1,78-2,59). El aclaramiento intercompartimental fue de 0,336 l/día (IC del 95 %: 0,261-0,443).
Metabolismo: El metabolismo de ENSPRYNG no se ha estudiado directamente, ya que los anticuerpos monoclonales se eliminan principalmente por catabolismo.
Eliminación: El aclaramiento total de ENSPRYNG depende de la concentración. Se calcula que el aclaramiento lineal (que representa aproximadamente la mitad del aclaramiento total en estado de equilibrio utilizando la dosis recomendada en los pacientes con NMO y TENMO) es de 0,0601 l/día (IC del 95 %: 0,0524-0,0695). La t1/2 terminal asociada es de aproximadamente 30 días (intervalo, 22-37 días) según los datos agrupados de los estudios de fase 3.
Farmacocinética en poblaciones especiales: Los análisis de farmacocinética poblacional en pacientes adultos con NMO o TENMO revelaron que la edad, el sexo y la raza no influyeron significativamente en la farmacocinética del satralizumab. Aunque el peso corporal influyó en la farmacocinética del satralizumab, no se recomienda ajustar la dosis en función de ninguna de estas características demográficas.
Población pediátrica: Los datos obtenidos en 8 pacientes adolescentes [13-17 años] que recibieron la pauta posológica utilizada en adultos demuestran que los parámetros de FC poblacional del satralizumab no son significativamente diferentes de los de la población adulta.
Por tanto, no es necesario ajustar la dosis.
Población geriátrica: No se han realizado estudios específicos para investigar la FC del satralizumab en pacientes mayores de 65 años; sin embargo, en los estudios clínicos BN40898 y BN40900 se incluyó a pacientes con NMO o TENMO de 65 a 74 años.
Los análisis de FC poblacional obtenidos a partir de los datos de estos pacientes revelaron que la edad no afectó a la FC del satralizumab.
Insuficiencia renal: No se ha realizado ningún estudio formal del efecto de la insuficiencia renal en la FC de satralizumab; no obstante, en los estudios clínicos BN40898 y BN40900 se incluyeron pacientes con insuficiencia renal leve (aclaramiento de creatinina <80 ml/min y ≥50 ml/min). Como era de esperar a tenor de los mecanismos conocidos de aclaramiento del satralizumab, la farmacocinética no resultó afectada en estos pacientes, por lo que no es necesario ajustar la dosis.
Insuficiencia hepática: No se ha realizado ningún estudio formal del efecto de la insuficiencia hepática en la FC del satralizumab.
CONTRAINDICACIONES: ENSPRYNG está contraindicado en pacientes con:
Hipersensibilidad conocida al satralizumab o a cualquiera de sus excipientes.
Infección activa por hepatitis B
Tuberculosis activa o latente no tratada.
INSTRUCCIONES ESPECIALES DE USO, MANIPULACIÓN Y ELIMINACIÓN: ENSPRYNG es para una sola dosis. No inyecte el medicamento si el líquido está turbio, ha cambiado de color o contiene partículas. Compruebe la JPC y el DSA en busca de daños. No los utilice si están agrietados o rotos.
Eliminación de la JPC y el DSA: Deben respetarse estrictamente los aspectos siguientes relativos al uso y la eliminación de la JPC y el DSA:
La JPC nunca debe reutilizarse.
Tire la jeringa usada en un recipiente para objetos punzantes inmediatamente después de su uso.
Deseche (tire) la JPC y el DSA de conformidad con los requisitos locales o siguiendo las indicaciones de su profesional sanitario.
Mantenga las JPC, los DSA y todos los medicamentos fuera del alcance de los niños.
Eliminación de medicamentos no utilizados o caducados: Se debe reducir al mínimo la liberación de productos farmacéuticos al medio ambiente. Los medicamentos no se deben tirar por los desagües y se debe evitar tirarlos a la basura. Utilice los «sistemas de recogida» establecidos si están disponibles en su zona.
PROPIEDADES Y EFECTOS FARMACOLÓGICOS: Propiedades farmacodinámicas: En estudios clínicos con ENSPRYNG en la NMO y los TENMO, se observaron disminuciones de la proteína C-reactiva (CRP), el fibrinógeno y el complemento (C3, C4 y CH50).
Mecanismo de acción: El satralizumab es un anticuerpo monoclonal (AcM) de tipo IgG2 humanizado que se une al receptor de la IL-6 humana (IL-6R) soluble y unido a la membrana, impidiendo así la señalización anterógrada de la IL-6 a través de estos receptores.
La IL-6 es una citocina pleótropa producida por varios tipos de células e interviene en diversos procesos inflamatorios, como la activación de los linfocitos B, la diferenciación de los linfocitos B en plasmoblastos y la producción de autoanticuerpos, la activación y diferenciación de los linfocitos Th17, la inhibición de los linfocitos T reguladores y los cambios de permeabilidad de la barrera hematoencefálica. Las concentraciones de IL-6 aumentan en el líquido cefalorraquídeo y el suero de los pacientes con NMO y TENMO durante los periodos de actividad de la enfermedad. Se han implicado algunas funciones de la IL-6 en la patogenia de la NMO y el TENMO, como la producción de autoanticuerpos patológicos contra la acuaporina 4 (AQP4), una proteína que actúa como canal de agua expresada principalmente por los astrocitos del SNC.
Estudios clínicos y de eficacia: La eficacia y la seguridad de ENSPRYNG se evaluaron en dos ensayos clínicos fundamentales de fase III (BN40898 y BN40900) en los que participaron pacientes con un diagnóstico de NMO seropositivo o seronegativo para IgG-AQP4 (criterios de Wingerchuk 2006) o de TENMO seropositivo para IgG-AQP4 (criterios de Wingerchuk 2007). Un análisis retrospectivo determinó que estos pacientes también cumplían los criterios más recientes propuestos por el grupo internacional para el diagnóstico de la NMO (Ref.: Wingerchuk et al 2015). El efecto de ENSPRYNG se estudió en pacientes adultos (estudios BN40898 y BN40900) y adolescentes (de ≥12 a <18 años) (estudio BN40898). La inclusión de pacientes adultos con NMO seronegativos para IgG-AQP4 se limitó aproximadamente al 30 % en ambos estudios para que la población del estudio reflejara la población de pacientes con NMO en la vida real.
El criterio de eficacia principal en ambos estudios fueron las recidivas definidas por el protocolo (RDP) basadas en un empeoramiento preespecificado de la Escala ampliada del estado de discapacidad (EDSS) y la Escala de estado funcional (FSS), confirmado por un Comité de Valoración Clínica (CVC) independiente. El análisis del criterio de valoración principal fue el tiempo transcurrido hasta la primera RDP confirmada por el CVC mediante una evaluación de la EDSS/FSS realizada en los 7 días siguientes a la notificación de los síntomas por el paciente (recidiva confirmada).
Estudio BN40898 (también conocido como SA-307JG o SAkuraSky): El estudio BN40898 fue un ensayo clínico aleatorizado, multicéntrico, con doble enmascaramiento y controlado con placebo para evaluar el efecto de ENSPRYNG en combinación con un TID estable (CO hasta 15 mg/día [equivalente de prednisolona], AZA hasta 3 mg/kg/día o MMF hasta 3000 mg/día; los adolescentes recibieron una combinación de AZA y CO o MMF y CO). El estudio incluyó 83 pacientes seropositivos y seronegativos para IgG-AQP4 (entre ellos, 7 adolescentes). Los pacientes recibieron las 3 primeras dosis únicas de 120 mg de ENSPRYNG o de un placebo equivalente mediante inyección s. c. en el abdomen o el muslo cada 2 semanas durante las 4 primeras semanas y, posteriormente, una vez cada 4 semanas.
En la tabla 2 se presentan el diseño del estudio y las características basales de la población del estudio.
El estudio se basó en los acontecimientos y el periodo de estudio con doble enmascaramiento para la evaluación de la eficacia finalizó cuando se alcanzó un total de 26 recidivas confirmadas.
|
Tabla 2. Diseño y características basales del estudio BN40898 |
||
|
Nombre del estudio |
Estudio BN40898 (N = 83) |
|
|
Diseño del estudio |
||
|
Población del estudio |
Pacientes adolescentes y adultos con NMO o TENMO, tratados con un TID estable Edad de 12-74 años, ≥2 recidivas en los 2 últimos años antes de la selección (con al menos una recidiva en los 12 meses previos a la selección), EDSS de 0 a 6,5 |
|
|
Duración del estudio para la evaluación de la eficacia |
Basada en acontecimientos (26 recidivas definidas por el protocolo confirmadas por el CVC) Mediana del tiempo de seguimiento: 100 semanas para ENSPRYNG, 74 semanas para el placebo |
|
|
Grupos de tratamiento, aleatorización 1:1 |
Grupo A: ENSPRYNG 120 mg s. c. Grupo B: placebo |
|
|
Características basales |
ENSPRYNG + TID (n = 41) |
Placebo + TID (n = 42) |
|
Diagnóstico, n (%): NMO TENMO |
33 (80,5) 8 (19,5) |
28 (66,7) 14 (33,3) |
|
Seropositividad para IgG-AQP4, n (%) |
27 (65,9) |
28 (66,7) |
|
Media de edad en años (DE) (mín.-máx.) |
40,8 (16,1) (13 - 73) |
43,4 (12,0) (14 - 65) |
|
Adolescentes (≥12 a <18 años), n (%) |
4 (9,8) |
3 (7,1) |
|
Distribución por sexos, n (%) varones/n (%) mujeres |
4 (9,8) / 37 (90,2) |
2 (4,8) / 40 (95,2) |
|
Tratamiento inmunodepresor (TID), n (%): Corticosteroides orales (CO) Azatioprina (AZA) Micofenolato mofetilo (MMF) AZA + CO* MMF + CO* |
17 (41,5) 16 (39,0) 4 (9,8) 3 (7,3) 1 (2,4) |
20 (47,6) 13 (31,0) 8 (19,0) 0 1 (2,4) |
|
* Combinación permitida en pacientes adolescentes. |
||
Estudio BN40900 (también conocido como SA-309JG o SAkuraStar): El estudio BN40900 fue un ensayo clínico aleatorizado, multicéntrico, con doble enmascaramiento y controlado con placebo para evaluar el efecto de ENSPRYNG en monoterapia en comparación con un placebo. El estudio incluyó 95 pacientes seropositivos y seronegativos para IgG-AQP4. Los pacientes recibieron las 3 primeras dosis únicas de 120 mg de ENSPRYNG o de un placebo equivalente mediante inyección s. c. en el abdomen o el muslo cada 2 semanas durante las 4 primeras semanas y, posteriormente, una vez cada 4 semanas.
En la tabla 3 se presentan el diseño del estudio y las características basales de la población del estudio.
El periodo del estudio con doble enmascaramiento para la evaluación de la eficacia finalizó 1,5 años después de la fecha de aleatorización del último paciente incluido.
|
Tabla 3 Diseño y características basales del estudio BN40900 |
||
|
Nombre del estudio |
Estudio BN40900 (N = 95) |
|
|
Diseño del estudio |
||
|
Población del estudio |
Pacientes adultos con NMO o TENMO Edad de 18-74 años, ≥1 recidiva o primera crisis en los últimos 12 meses antes de la selección, EDSS de 0 a 6,5. Los pacientes habían recibido tratamiento previo para prevenir las recidivas del TENMO o no habían recibido tratamiento previo. |
|
|
Duración del estudio para la evaluación de la eficacia |
Basada en acontecimientos (44 recidivas definidas por el protocolo confirmadas por el CVC o 1,5 años después de la fecha de aleatorización del último paciente incluido, lo que ocurra antes) Mediana del tiempo de seguimiento: 95,4 semanas para ENSPRYNG, 60,5 semanas para el placebo |
|
|
Grupos de tratamiento, aleatorización 2:1 |
Monoterapia: Grupo A: ENSPRYNG 120 mg s. c. Grupo B: placebo |
|
|
Características basales |
ENSPRYNG (n = 63) |
Placebo (n = 32) |
|
Diagnóstico, n (%): NMO TENMO |
47 (74,6) 16 (25,4) |
24 (75,0) 8 (25,0) |
|
Seropositividad para IgG-AQP4, n (%) |
41 (65,1) |
23 (71,9) |
|
Media de edad en años (DE) (mín.-máx.) |
45,3 (12,0) (21 - 70) |
40,5 (10,5) (20 - 56) |
|
Distribución por sexos, n (%) varones/n (%) mujeres |
17 (27,0) / 46 (73,0) |
1 (3,1) / 31 (96,9) |
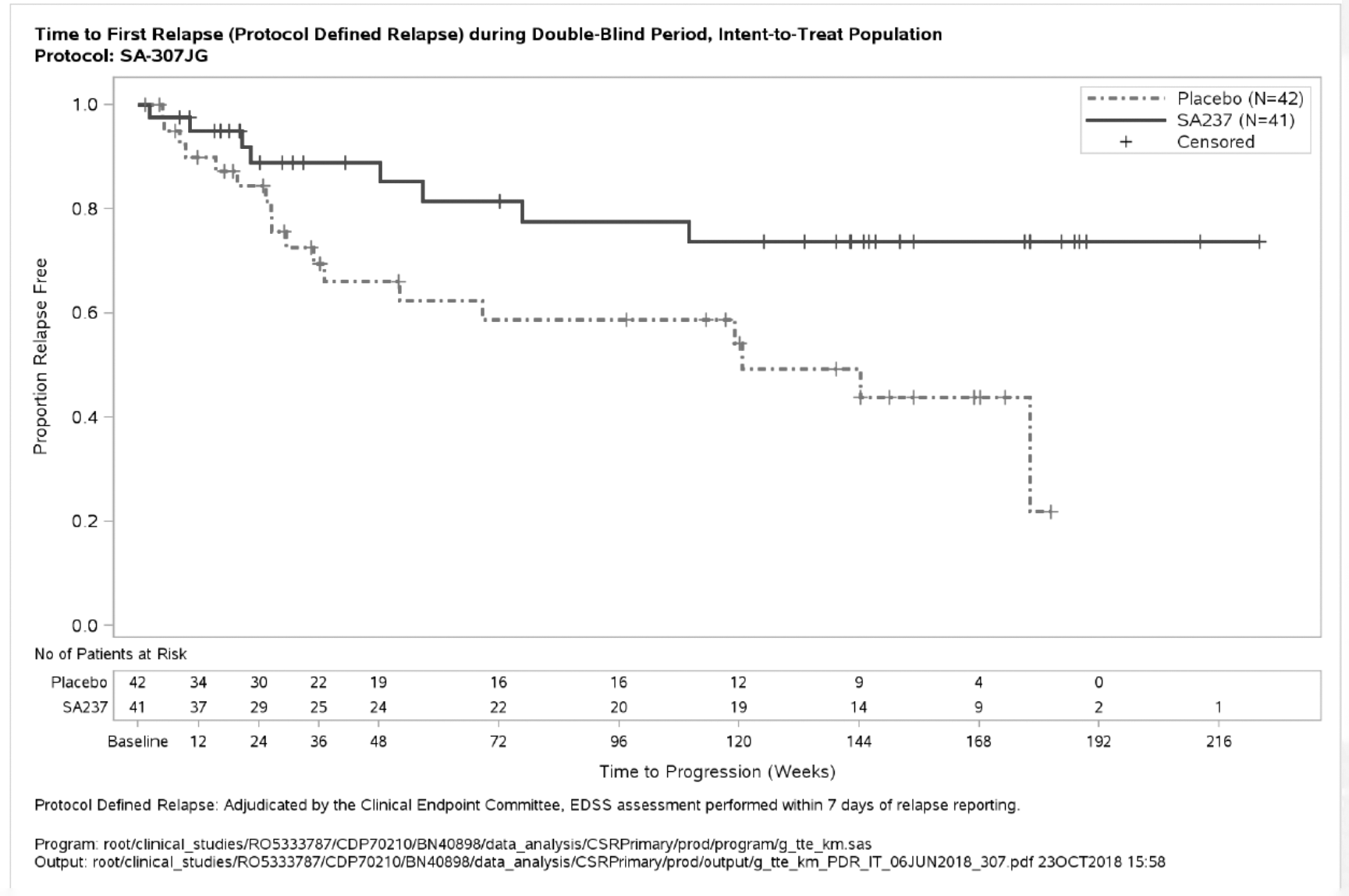
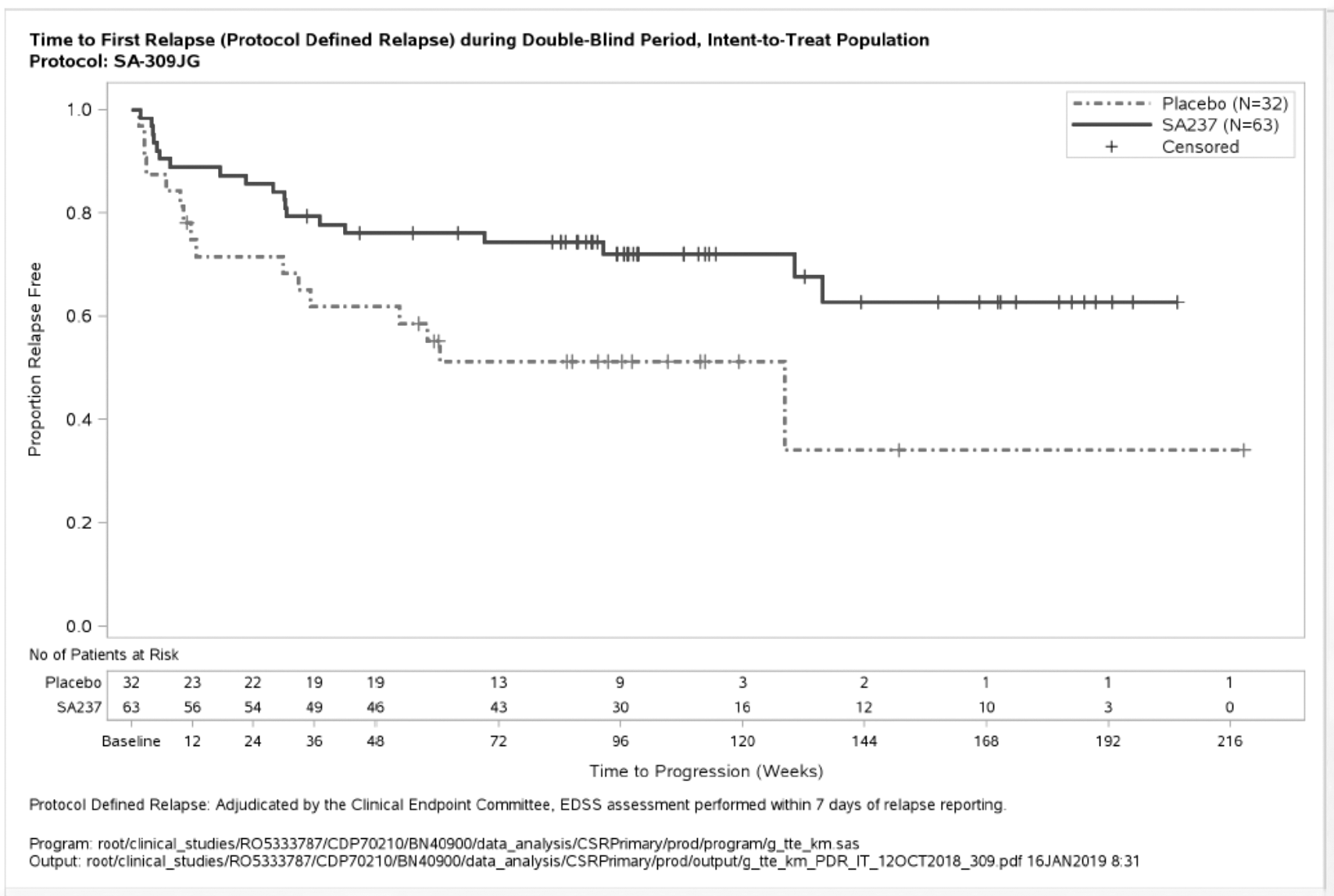
Criterio de valoración principal de la eficacia: El tratamiento con ENSPRYNG produjo una reducción estadísticamente significativa, del 62 %, del riesgo de sufrir una recaída confirmada (razón de riesgos instantáneos [hazard ratio, HR] [IC del 95 %]: 0,38 [0,16-0,88]; p [orden logarítmico] = 0,0184) cuando se administró en combinación con un TID estable (estudio BN40898) y una reducción del 55 % del riesgo de recidiva confirmada (HR [IC del 95 %]: 0,45 [0,23-0,89]; p [orden logarítmico] = 0,0184) cuando se utilizó en monoterapia (estudio BN40900) en comparación con el placebo. A las 48 semanas, el 88,9 % y el 76,1 % de los pacientes tratados con ENSPRYNG en combinación con TID o en monoterapia seguían sin recidivas confirmadas, respectivamente. A las 96 semanas, el 77,6 % y el 72,1 % de los pacientes tratados con ENSPRYNG en combinación con TID o en monoterapia seguían sin recidivas confirmadas, respectivamente. Cuando se agruparon los datos de los dos estudios, se observó una reducción del 58 % del riesgo de recidiva confirmada con el tratamiento con ENSPRYNG en comparación con el placebo (HR [IC del 95 %]: 0,42 [0,25-0,71]; p [orden logarítmico] = 0,0008) (véase la tabla 4, figura 1, figura 2).
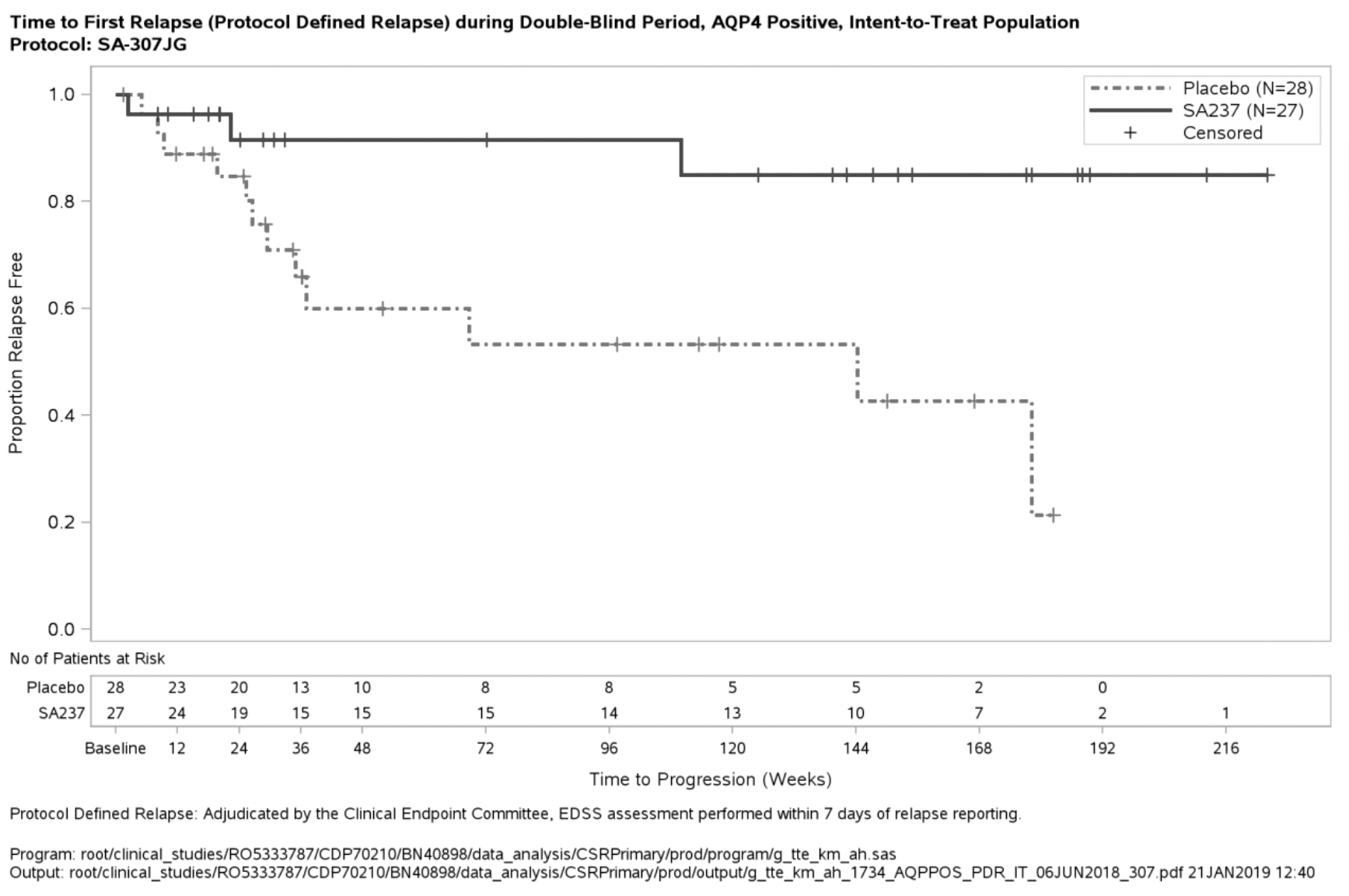
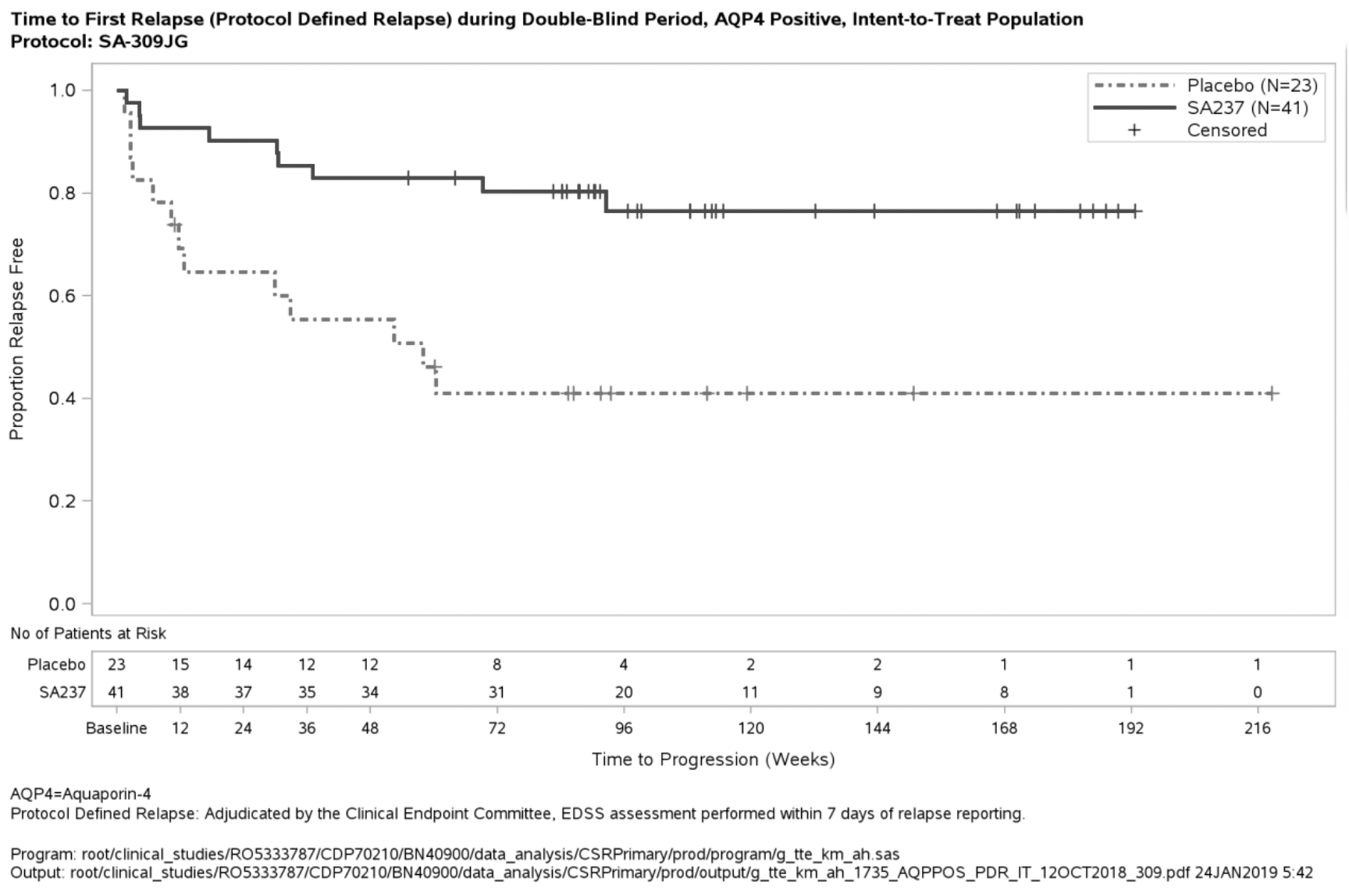
El efecto más potente en un subgrupo se observó en los pacientes seropositivos para IgG-AQP4. En los pacientes seropositivos para IgG-AQP4, el riesgo relativo de sufrir una recidiva confirmada se redujo en un 79 % en el estudio BN40898 (HR [IC del 95 %]: 0,21 [0,06-0,75]) y en un 74 % en el estudio BN40900 (HR[IC del 95 %]: 0,26 [0,11-0,63]). A las 48 semanas, permanecían sin recidivas confirmadas el 91,5 % y el 82,9 % de los pacientes seropositivos para IgG-AQP4 tratados con ENSPRYNG en combinación con TID o en monoterapia, respectivamente. A las 96 semanas, permanecían sin recidivas confirmadas el 91,5 % y el 76,5 % de los pacientes seropositivos para IgG-AQP4 tratados con ENSPRYNG en combinación con TID o en monoterapia, respectivamente. Cuando se agruparon los datos de los estudios BN40898 y BN40900, se observó que el tratamiento con ENSPRYNG, con o sin TID, lograba una reducción global del riesgo del 75 % (HR [IC del 95 %]; 0,25 [0,12-0,50]) en los pacientes seropositivos para IgG-AQP4 (véase la tabla 4, figura 3, figura 4). En cuanto a los pacientes seronegativos para IgG-AQP4, las diferencias en el tiempo hasta la primera recidiva confirmada entre los pacientes tratados con ENSPRYNG con o sin TID y los tratados con un placebo con o sin TID no fueron significativas (BN40898 y BN40900 agrupados: HR [IC del 95 %]: 0,97 [0,41-2,33]).
|
Tabla 4. Criterios de valoración de la eficacia fundamentales de los estudios BN40898 y BN40900 |
||||
|
BN40898 |
BN40900 |
|||
|
ENSPRYNG + TID |
Placebo + TID |
ENSPRYNG |
Placebo |
|
|
(n = 41) |
(n = 42) |
(n = 63) |
(n = 32) |
|
|
Criterio de valoración principal |
||||
|
Reducción del riesgo (estudios individuales) |
62 % (HR: 0,38; IC del 95 %: 0,16, 0,88; p = 0,0184) |
55 % (HR: 0,45; IC del 95 %: 0,23, 0,89; p = 0,0184) |
||
|
Reducción del riesgo (análisis agrupado) |
58 % (HR: 0,42; IC del 95 %: 0,25, 0,71; p = 0,0008) |
|||
|
Proporción de pacientes sin recidivas confirmadas a las 48 semanas |
88,9 % (IC del 95 %: 72,81, 95,70) |
66,0 % (IC del 95 %: 47,65, 79,25) |
76,1 % (IC del 95 %: 63,55, 84,86) |
61,9 % (IC del 95 %: 42,66, 76,26) |
|
Proporción de pacientes sin recidivas confirmadas a las 96 semanas |
77,6 % (IC del 95 %: 58,08, 88,82) |
58,7 % (IC del 95 %: 39,85, 73,43) |
72,1 % (IC del 95 %: 58,91, 81,75) |
51,2 % (IC del 95 %: 32,36, 67,23) |
|
Análisis por subgrupos del criterio de valoración principal (pacientes seropositivos para IgG-AQP4) |
||||
|
Número de pacientes seropositivos para IgG-AQP4 (n) |
27 |
28 |
41 |
23 |
|
Reducción del riesgo (estudios individuales) |
79 % (HR: 0,21; IC del 95 %: 0,06, 0,75; p = 0,0086) |
74 % (HR: 0,26; IC del 95 %: 0,11, 0,63; p = 0,0014) |
||
|
Reducción del riesgo (análisis agrupado) |
75 % (HR: 0,25; IC del 95 %: 0,12, 0,50; p <0,0001) |
|||
|
Proporción de pacientes sin recidivas confirmadas a las 48 semanas |
91,5 % (IC del 95 %: 69,64, 97,83) |
59,9 % (IC del 95 %: 36,25, 77,25) |
82,9 % (IC del 95 %: 67,49, 91,47) |
55,4 % (IC del 95 %: 32,96, 73,08) |
|
Proporción de pacientes sin recidivas confirmadas a las 96 semanas |
91,5 % (IC del 95 %: 69,64, 97,83) |
53,3 % (IC del 95 %: 29,34, 72,38) |
76,5 % (IC del 95 %: 59,22, 87,21) |
41,1 % (IC del 95 %: 20,76, 60,41) |
Figura 1. Estudio BN40898: tiempo hasta la primera recidiva confirmada durante el periodo de doble enmascaramiento (población IDT)
|
Time to First Relapse (Protocol Defined Relapse) during Double-Blind Period, Intent-to-Treat Population |
Tiempo hasta la primera recidiva (recidiva definida por el protocolo) durante el periodo de doble enmascaramiento, población por intención de tratar |
|
Protocol |
Protocolo |
|
Placebo |
Placebo |
|
Censored |
Censurados |
|
Proportion Relapse Free |
Proporción sin recidivas |
|
No of Patients at Risk |
N.º de pacientes en riesgo |
|
Baseline |
Momento basal |
|
Time to Progression (Weeks) |
Tiempo hasta la progresión (semanas) |
|
Protocol Defined Relapse: Adjudicated by the Clinical Endpoint Committee, EDSS assessment performed within 7 days of relapse reporting |
Recidiva definida en el protocolo: confirmada por el Comité de Valoración Clínica, evaluación EDSS realizada en los 7 días siguientes a la notificación de la recidiva |
|
Program |
Programa |
|
Output |
Informe |
Figura 2. Estudio BN40900: tiempo hasta la primera recidiva confirmada durante el periodo de doble enmascaramiento (población IDT)
|
Time to First Relapse (Protocol Defined Relapse) during Double-Blind Period, Intent-to-Treat Population |
Tiempo hasta la primera recidiva (recidiva definida por el protocolo) durante el periodo de doble enmascaramiento, población por intención de tratar |
|
Protocol |
Protocolo |
|
Placebo |
Placebo |
|
Censored |
Censurados |
|
Proportion Relapse Free |
Proporción sin recidivas |
|
No of Patients at Risk |
N.º de pacientes en riesgo |
|
Baseline |
Momento basal |
|
Time to Progression (Weeks) |
Tiempo hasta la progresión (semanas) |
|
Protocol Defined Relapse: Adjudicated by the Clinical Endpoint Committee, EDSS assessment performed within 7 days of relapse reporting |
Recidiva definida en el protocolo: confirmada por el Comité de Valoración Clínica, evaluación EDSS realizada en los 7 días siguientes a la notificación de la recidiva |
|
Program |
Programa |
|
Output |
Informe |
Figura 3. Estudio BN40898: tiempo hasta la primera recidiva confirmada durante el periodo de doble enmascaramiento en los pacientes seropositivos para IgG-AQP4
|
Time to First Relapse (Protocol Defined Relapse) during Double-Blind Period, AQP4 Positive, Intent-to-Treat Population |
Tiempo hasta la primera recidiva (recidiva definida en el protocolo) durante el periodo de doble enmascaramiento, pacientes positivos para AQP4, población por intención de tratar |
|
Protocol |
Protocolo |
|
Placebo |
Placebo |
|
Censored |
Censurados |
|
Proportion Relapse Free |
Proporción sin recidivas |
|
No of Patients at Risk |
N.º de pacientes en riesgo |
|
Baseline |
Momento basal |
|
Time to Progression (Weeks) |
Tiempo hasta la progresión (semanas) |
|
Protocol Defined Relapse: Adjudicated by the Clinical Endpoint Committee, EDSS assessment performed within 7 days of relapse reporting |
Recidiva definida en el protocolo: confirmada por el Comité de Valoración Clínica, evaluación EDSS realizada en los 7 días siguientes a la notificación de la recidiva |
|
Program |
Programa |
|
Output |
Informe |
Figura 4. Estudio BN40900: tiempo hasta la primera recidiva confirmada durante el periodo de doble enmascaramiento en los pacientes seropositivos para IgG-AQP4
|
Time to First Relapse (Protocol Defined Relapse) during Double-Blind Period, AQP4 Positive, Intent-to-Treat Population |
Tiempo hasta la primera recidiva (recidiva definida en el protocolo) durante el periodo de doble enmascaramiento, pacientes positivos para AQP4, población por intención de tratar |
|
Protocol |
Protocolo |
|
Placebo |
Placebo |
|
Censored |
Censurados |
|
Proportion Relapse Free |
Proporción sin recidivas |
|
No of Patients at Risk |
N.º de pacientes en riesgo |
|
Baseline |
Momento basal |
|
Time to Progression (Weeks) |
Tiempo hasta la progresión (semanas) |
|
AQP4 = Aquaporin-4 |
AQP4 = acuaporina 4 |
|
Protocol Defined Relapse: Adjudicated by the Clinical Endpoint Committee, EDSS assessment performed within 7 days of relapse reporting |
Recidiva definida en el protocolo: confirmada por el Comité de Valoración Clínica, evaluación EDSS realizada en los 7 días siguientes a la notificación de la recidiva |
|
Program |
Programa |
|
Output |
Informe |
Características basales y eficacia en pacientes adolescentes (estudio BN40898): La media de edad de los 7 pacientes adolescentes incluidos durante el periodo de doble enmascaramiento del estudio BN40898 era de 15,4 años y la mediana del peso corporal, de 79,6 kg. La mayoría de los pacientes adolescentes eran mujeres (n = 6). Cuatro pacientes eran de raza blanca; 2, de raza negra/afroamericanos y 1, de raza asiática. Tres de los 7 (42,9 %) pacientes adolescentes eran seropositivos para IgG-AQP4 en el momento de la selección (2 del grupo de placebo y 1 del grupo de ENSPRYNG). Durante el periodo de doble enmascaramiento, 1 de los 3 adolescentes del grupo de placebo y 1 de los 4 del grupo de ENSPRYNG tuvieron una recidiva confirmada. Debido al pequeño tamaño de la muestra, no se calculó la razón de riesgos instantáneos para el criterio de valoración principal del tiempo hasta la primera recidiva confirmada en este subgrupo.
Inmunogenia: En el estudio de fase III BN40898 (combinación con TID) y en el estudio de fase III BN40900 (monoterapia) se observaron anticuerpos antiterapéuticos (AcAT) en el 41 % y el 71 % de los pacientes tratados con ENSPRYNG en el periodo de doble enmascaramiento, respectivamente. Se desconoce la capacidad de estos AcAT para neutralizar la unión de ENSPRYNG.
La exposición fue menor en los pacientes con AcAT, pero no se observó ningún efecto de los AcAT en la seguridad ni un efecto claro en la eficacia ni los marcadores farmacodinámicos indicativos de unión a la diana.
El tratamiento con satralizumab produjo una reducción similar del riesgo de experimentar una recidiva confirmada en los pacientes de los estudios de fase III a pesar de las diferentes tasas de AcAT observadas entre estos estudios. Los pacientes con un mayor peso corporal y una menor exposición tuvieron más probabilidades de presentar AcAT (con independencia del tratamiento de base con TID), si bien el efecto del tratamiento fue similar en todos los grupos de peso corporal, ya se utilizase en combinación con TID o en monoterapia. La dosis recomendada es adecuada para todos los pacientes y no está justificada la interrupción del tratamiento ni la modificación de la dosis en los pacientes que presenten AcAT.
REACCIONES ADVERSAS: Ensayos clínicos: Resumen del perfil de seguridad: La seguridad de ENSPRYNG en monoterapia o en combinación con TID se evaluó a partir de los datos de dos ensayos clínicos de fase III aleatorizados, multicéntricos, con doble enmascaramiento y controlados con placebo (BN40898 y BN40900), en los que participaron 63 pacientes expuestos a ENSPRYNG en monoterapia y 41 expuestos a ENSPRYNG en combinación con un TID (v. Estudios clínicos y de eficacia).
Las reacciones adversas al medicamento (RAM) notificadas con más frecuencia fueron cefalea, artralgia y reacciones relacionadas con la inyección.
Resumen tabulado de las reacciones adversas descritas en los ensayos clínicos: En la tabla 1 se resumen las reacciones adversas que se han notificado en relación con el uso de ENSPRYNG en monoterapia o en combinación con TID en los ensayos clínicos. El periodo de tratamiento fue más prolongado en los pacientes de los grupos tratados con ENSPRYNG en ambos estudios clínicos que en los grupos de placebo (o de placebo en combinación con TID); las reacciones adversas se evaluaron durante 194 años-paciente (AP) en los grupos de ENSPRYNG y 100 AP en los grupos de placebo. Las reacciones adversas notificadas en los ensayos clínicos (tabla 1) se enumeran por clase de órgano, aparato o sistema del MedDRA.
La categoría de frecuencia correspondiente a cada reacción adversa se basa en la convención siguiente: muy frecuente (≥1/10), frecuente (≥1/100 a <1/10), poco frecuente (≥1/1000 a <1/100), rara (≥1/10 000 a <1/1000), muy rara (<1/10 000).
|
Tabla 1: Resumen de las reacciones adversas observadas en pacientes tratados con ENSPRYNG en monoterapia o en combinación con tratamiento inmunodepresor en los ensayos clínicos |
|||||
|
Reacciones adversas (MedDRA) |
Episodios por 100 AP |
Número de pacientes (%) |
Categoría de frecuencia con ENSPRYNG |
||
|
ENSPRYNG AP = 193,74 |
Placebo1 AP = 100,10 |
ENSPRYNG n = 104 |
Placebo1 n = 74 |
||
|
Trastornos del sistema nervioso |
|||||
|
Cefalea |
18,07 |
10,99 |
20 (19,2 %) |
8 (10,8 %) |
Muy frecuente |
|
Migraña |
2,06 |
0,00 |
4 (3,8 %) |
0 |
Frecuente |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
|||||
|
Reacciones relacionadas con la inyección |
17,03 |
8,99 |
13 (12,5 %) |
7 (9,5 %) |
Muy frecuente |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
|||||
|
Artralgia |
7,23 |
1,0 |
14 (13,5 %) |
1 (1,4 %) |
Muy frecuente |
|
Rigidez musculoesquelética |
2,58 |
0,00 |
5 (4,8 %) |
0 |
Frecuente |
|
Trastornos de la piel y del tejido subcutáneo |
|||||
|
Exantema |
7,23 |
4,00 |
9 (8,7 %) |
3 (4,1 %) |
Frecuente |
|
Prurito |
4,13 |
1,00 |
6 (5,8 %) |
1 (1,4 %) |
Frecuente |
|
Trastornos psiquiátricos |
|||||
|
Insomnio |
3,10 |
1,00 |
6 (5,8 %) |
1 (1,4 %) |
Frecuente |
|
Trastornos generales y alteraciones en el lugar de administración |
|||||
|
Edema periférico |
2,58 |
0,00 |
5 (4,8 %) |
0 |
Frecuente |
|
Trastornos respiratorios, torácicos y mediastínicos |
|||||
|
Rinitis alérgica |
2,06 |
0,00 |
4 (3,8 %) |
0 |
Frecuente |
|
Trastornos de la sangre y del sistema linfático |
|||||
|
Hipofibrinogenemia |
1,55 |
0,00 |
3 (2,9 %) |
0 |
Frecuente |
|
1 Placebo o placebo en combinación con TID |
|||||
Descripción de determinadas reacciones adversas descritas en los ensayos clínicos: Reacciones relacionadas con la inyección (RRI): Las RRI notificadas en pacientes tratados con ENSPRYNG en monoterapia o en combinación con TID fueron predominantemente leves o moderadas y la mayoría aparecieron en las 24 horas siguientes a la inyección. Los síntomas sistémicos notificados con más frecuencia fueron diarrea y cefalea. Las reacciones locales en el lugar de inyección más frecuentes consistieron en rubefacción, eritema, prurito, exantema y dolor. Ninguna de las reacciones relacionadas con la inyección obligó a interrumpir la administración o suspender definitivamente el tratamiento.
Infecciones: En el estudio de ENSPRYNG en monoterapia, la tasa de infecciones fue inferior entre los pacientes tratados con ENSPRYNG [99,8 episodios/100 AP (IC del 95 %: 82,4, 119,8)] que entre los tratados con placebo [162,6 episodios/100 AP (IC del 95 %: 125,8, 206,9)]. La tasa de infecciones graves fue de 5,2 episodios/100 AP (IC del 95 %: 1,9, 11,3) entre los pacientes tratados con ENSPRYNG, en comparación con 9,9 episodios/100 AP (IC del 95 %: 2,7, 25,2) entre los que recibieron el placebo.
Entre los pacientes tratados con ENSPRYNG en combinación con TID, la tasa de infecciones fue de 132,5 episodios/100 AP (IC del 95 %: 108,2, 160,5) en comparación con 149,6 episodios/100 AP (IC del 95 %: 120,1, 184,1) en los pacientes tratados con un placebo en combinación con TID; la tasa de infecciones graves fue de 2,6 episodios/100 AP (IC del 95 %: 0,3, 9,2) en comparación con 5,0 episodios/100 AP (IC del 95 %: 1,0, 14,7) en los pacientes que recibieron el placebo en combinación con TID.
Anomalías analíticas: Neutrófilos: En el periodo de tratamiento con doble enmascaramiento, se observó una disminución de los neutrófilos en el 31,7 % de los pacientes tratados con ENSPRYNG (en monoterapia o en combinación con TID), en comparación con el 21,6 % de los pacientes que recibieron el placebo (con o sin TID). La mayoría de las disminuciones de los neutrófilos fueron transitorias o intermitentes.
De los pacientes del grupo de ENSPRYNG, el 9,6 % tenía un valor de neutrófilos por debajo de 1 x 109/l, en comparación con el 5,4 % de los que recibieron el placebo con o sin TID; el descenso de los neutrófilos no se asoció temporalmente a ninguna infección grave.
Plaquetas: En el periodo de tratamiento con doble enmascaramiento, se produjo una disminución de la cifra de plaquetas en el 24,0 % de los pacientes tratados con ENSPRYNG (en monoterapia o en combinación con TID) en comparación con el 9,5 % de los pacientes que recibieron un placebo con o sin TID. La disminución de la cifra de plaquetas no se asoció a episodios hemorrágicos.
La mayoría de los descensos de las plaquetas fueron transitorios y los valores no disminuyeron por debajo de 75 × 109/l. Ninguno de los pacientes presentó una disminución de la cifra de plaquetas hasta valores ≤50 × 109/l.
Enzimas hepáticas: En el periodo de tratamiento con doble enmascaramiento, se produjeron elevaciones de la ALT o la AST en el 27,9 % y el 18,3 % de los pacientes tratados con ENSPRYNG (en monoterapia o en combinación con TID), respectivamente, en comparación con el 12,2 % y el 13,5 % de los pacientes que recibieron un placebo o un placebo más TID. La mayoría de las elevaciones fueron inferiores a 3 veces el LSN, fueron transitorias y se resolvieron sin necesidad de interrumpir la administración de ENSPRYNG.
Se produjeron elevaciones de la ALT o la AST >3 veces LSN en el 2,9 % y el 1,9 % de los pacientes tratados con ENSPRYNG (en monoterapia o en combinación con IST), respectivamente, que no se asociaron a aumentos de la bilirrubina total. En un paciente tratado con ENSPRYNG en combinación con TID se observó una elevación de la ALT superior a 5 veces el LSN cuatro semanas después del inicio del tratamiento, que se normalizó tras la suspensión de ENSPRYNG.
Parámetros lipídicos: En el periodo de tratamiento con doble enmascaramiento, el 10,6 % de los pacientes tratados con ENSPRYNG (en monoterapia o en combinación con TID) presentaron elevaciones del colesterol total superiores a 7,75mmol/l, en comparación con el 1,4 % de los pacientes que recibieron un placebo con o sin TID; el 18,3 % de los pacientes que recibieron ENSPRYNG presentaron elevaciones de los triglicéridos superiores a 3,42 mmol/l, en comparación con el 6,8 % de los pacientes que recibieron un placebo. Las elevaciones de los parámetros lipídicos no obligaron a interrumpir el tratamiento.
Experiencia posterior a la comercialización: No procede
INTERACCIONES CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN: No se han realizado estudios formales de interacciones farmacológicas con ENSPRYNG.
Los análisis de FC poblacional no detectaron ningún efecto de la AZA, los corticosteroides ni el MMF en el aclaramiento de ENSPRYNG.
Se ha investigado la capacidad del tratamiento con ENSPRYNG para reducir la exposición a medicamentos concomitantes metabolizados por las isoenzimas del CYP450 mediante el bloqueo de la señalización de la IL-6 utilizando modelos farmacocinéticos de base fisiológica (FCBF).
Esto indica que la supresión de la señalización de la IL-6 producida por el tratamiento con ENSPRYNG a partir de los valores basales bajos observados en los estudios de fase III tendrá solo un efecto escaso en la exposición a una serie de sustratos del CYP450 investigados (aumento ≤15 % del ABC con todos los sustratos de las isoenzimas del CYP 1A2, 3A4, 2D6 y 2C19). Por consiguiente, el riesgo de interacciones farmacológicas es bajo, aunque es necesario actuar con precaución cuando se administre ENSPRYNG o se suspenda su uso en pacientes que también estén recibiendo sustratos del CYP450 con un índice terapéutico estrecho.
ADVERTENCIAS Y PRECAUCIONES: Se observaron disminuciones en el recuento de neutrófilos en pacientes tratados con ENSPRYNG con una mayor incidencia que con el placebo. Los recuentos de neutrófilos deben controlarse de 4 a 8 semanas después del inicio del tratamiento y, posteriormente, al intervalo regular determinado clínicamente.
Generalidades: Con el fin de mejorar la trazabilidad de los medicamentos biológicos, debe registrarse claramente el nombre comercial y el número de lote del producto administrado.
Infecciones: Se debe retrasar la administración de ENSPRYNG en los pacientes que presenten una infección activa hasta que se haya resuelto (v. Posología y administración, Dosis aplazadas u omitidas).
Vacunas: No deben administrarse vacunas de microorganismos vivos o vivos atenuados simultáneamente con ENSPRYNG, ya que no se ha determinado su seguridad clínica. El intervalo entre la administración de vacunas de microorganismos vivos y el inicio del tratamiento con ENSPRYNG debe ser conforme a las directrices de vacunación actuales referentes a los inmunomoduladores o inmunodepresores.
No se dispone de datos sobre los efectos de la vacunación en pacientes tratados con ENSPRYNG. Se recomienda actualizar todas las vacunaciones de los pacientes de acuerdo con las directrices de vacunación actuales antes de iniciar el tratamiento con ENSPRYNG.
Enzimas hepáticas: Se han observado elevaciones leves y moderadas de las transaminasas hepáticas asociadas al tratamiento con ENSPRYNG; la mayoría de las elevaciones fueron inferiores a 5 veces el LSN, no limitaron el tratamiento y se resolvieron durante la administración de ENSPRYNG.
Las concentraciones de ALT y AST deben vigilarse cada 4 semanas durante los 3 primeros meses de tratamiento, luego cada 3 meses durante 1 año y, posteriormente, cuando esté clínicamente indicado. Las recomendaciones para la suspensión del tratamiento se presentan en el apartado Posología y administración, Modificaciones de la dosis.
Abuso y dependencia de fármacos: No se han realizado estudios sobre abuso y dependencia con este fármaco. Sin embargo, no hay indicios de que el tratamiento con ENSPRYNG produzca dependencia a partir de los datos disponibles.
Capacidad para conducir y utilizar máquinas: No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas. No obstante, no hay indicios de que el tratamiento con ENSPRYNG afecte a la capacidad para conducir y utilizar máquinas a partir de los datos disponibles.
POSOLOGÍA Y ADMINISTRACIÓN: Generalidades: La sustitución por cualquier otro medicamento biológico requiere el consentimiento del médico prescriptor.
No se han establecido la seguridad ni la eficacia de alternar o cambiar ENSPRYNG por productos que sean biosimilares pero que no se consideren intercambiables. Por consiguiente, ha de sopesarse cuidadosamente la relación riesgo-beneficio de alternar los medicamentos o cambiar de tratamiento.
Para evitar errores de medicación, es importante comprobar la etiqueta de la jeringa precargada para asegurarse de que el fármaco que se va a administrar es ENSPRYNG.
Posología recomendada: ENSPRYNG debe administrarse en forma de inyección subcutánea.
ENSPRYNG puede utilizarse en monoterapia o en combinación con corticosteroides orales (CO), azatioprina (AZA) o micofenolato mofetilo (MMF) (v. Estudios clínicos y de eficacia). Consulte también la ficha técnica completa de estos productos.
Dosis de carga: La dosis de carga recomendada es de 120 mg mediante inyección s. c. cada 2 semanas (primera dosis en la semana 0, segunda dosis en la semana 2 y tercera dosis en la semana 4) en las tres primeras administraciones.
Dosis de mantenimiento: La dosis de mantenimiento recomendada es de 120 mg mediante inyección s. c. cada 4 semanas.
Forma de administración: Los lugares de inyección recomendados son el abdomen y el muslo. Los lugares de inyección deben alternarse y las inyecciones nunca deben administrarse en lunares, cicatrices ni zonas donde la piel esté sensible, magullada, enrojecida, dura o con heridas.
En las instrucciones de uso se facilitan indicaciones detalladas para la administración de ENSPRYNG.
La primera inyección debe administrarse bajo la supervisión de un profesional sanitario cualificado. Un paciente adulto/cuidador puede administrar ENSPRYNG en el domicilio si el médico responsable del tratamiento lo considera apropiado y el paciente adulto/cuidador domina la técnica de inyección.
Los pacientes/cuidadores deben acudir al médico de inmediato si el paciente presenta síntomas de una reacción alérgica grave y consultar al profesional sanitario responsable si puede continuarse o no el tratamiento con ENSPRYNG.
Duración del tratamiento: ENSPRYNG está indicado para el tratamiento a largo plazo.
Dosis aplazadas u omitidas: Si se omite una inyección, deberá administrarse lo antes posible; no espere hasta la siguiente dosis prevista. Una vez administrada la dosis aplazada u omitida, deberá mantenerse el intervalo de 2 semanas (periodo de carga) o 4 semanas (periodo de mantenimiento) entre las dosis.
Modificaciones de la dosis
Anomalías de las enzimas hepáticas: Si la elevación de la alanina-transaminasa (ALT) o la aspartato-transaminasa (AST) es >5 veces el límite superior de la normalidad (LSN) y se asocia a cualquier grado de elevación de la bilirrubina, deberá suspenderse definitivamente el tratamiento con ENSPRYNG.
Si la elevación de la ALT o de la AST es >5 veces el LSN y no se asocia a elevación de la bilirrubina, debe interrumpirse el tratamiento con ENSPRYNG, que podrá reanudarse (inyección s. c. de 120 mg cada 4 semanas) cuando los valores de ALT y AST se hayan normalizado y en función de la evaluación de la relación riesgo-beneficio del tratamiento en el paciente. Si se toma la decisión de reanudar el tratamiento, se vigilarán estrechamente los parámetros hepáticos y, si se observa cualquier aumento posterior de la ALT/AST o la bilirrubina, se suspenderá definitivamente la administración del fármaco.
Instrucciones posológicas especiales:
Uso en pediatría: No se han estudiado la seguridad ni la eficacia de ENSPRYNG en la población pediátrica de menos de 18 años.
Uso en ancianos: No es necesario ajustar la dosis en los pacientes mayores de 65 años (v. Uso en ancianos y Farmacocinética en poblaciones especiales).
Insuficiencia renal: No se han estudiado formalmente la seguridad ni la eficacia de ENSPRYNG en pacientes con insuficiencia renal; sin embargo, no es previsible que sea necesario ajustar la dosis en los pacientes con insuficiencia renal (v. Insuficiencia renal y Farmacocinética en poblaciones especiales).
Insuficiencia hepática: No se han estudiado la seguridad ni la eficacia de ENSPRYNG en pacientes con insuficiencia hepática (v. Insuficiencia hepática y Farmacocinética en poblaciones especiales).
Otras poblaciones de pacientes especiales: No procede
SEGURIDAD PRECLÍNICA: Carcinogénesis: No se han realizado estudios de carcinogénesis en roedores para determinar el potencial carcinógeno del satralizumab. No se han observado lesiones proliferativas en un estudio de toxicidad crónica de 6 meses en macacos cangrejeros.
Genotoxicidad: No se han realizado estudios para determinar el potencial mutágeno del satralizumab.
No es previsible que los anticuerpos causen efectos sobre el ADN.
Deterioro de la fertilidad: No se observaron efectos sobre los órganos reproductores masculinos ni femeninos con el tratamiento crónico con satralizumab en monos.
Toxicidad para la reproducción: El tratamiento prenatal y posnatal con hasta 50 mg/kg/semana de satralizumab en monas gestantes y en sus crías no provocó efectos adversos en las madres, el desarrollo fetal, el desenlace de la gestación ni la supervivencia y desarrollo de las crías, incluida la capacidad de aprendizaje.
Las concentraciones de satralizumab en la leche materna fueron muy bajas (<0,9 % de las concentraciones plasmáticas maternas correspondientes).
Otros
Toxicidad con dosis múltiples: Los estudios preclínicos con monos, una especie con respuesta y reactividad cruzada al satralizumab, no revelaron riesgos especiales para el ser humano a tenor de los criterios de valoración de la seguridad farmacológica y de toxicidad con dosis únicas y múltiples. Cuando se administraron hasta 50 mg/kg de satralizumab a macacos cangrejeros una vez a la semana en estudios de toxicidad con dosis múltiples s. c. de 4 y 26 semanas de duración, no se observaron cambios en la toxicidad que se consideraran causados por la administración del fármaco. El único cambio de interés en estos estudios fue el aumento de la concentración sanguínea de IL-6, que se consideró el resultado de la acción farmacológica (acción neutralizante del IL-6R) del satralizumab, y no se asoció a hallazgos adversos. El tratamiento con satralizumab indujo una respuesta inmunitaria con anticuerpos antiterapéuticos en la mayoría de los animales tratados, que, sin embargo, no afectó a la respuesta farmacológica y no produjo eventos adversos.
Tolerabilidad local: La inyección s. c. de la formulación clínica de satralizumab no provocó ninguna reacción adversa en el lugar de administración en monos.
Reactividad tisular cruzada: La reactividad tisular cruzada detectada con satralizumab en tejidos de mono y ser humano refleja los lugares de expresión del IL-6R. No se detectó reactividad tisular cruzada de interés en otros tejidos.
Síndrome de liberación de citocinas: A tenor de los estudios in vitro realizados con sangre humana, el riesgo de liberación de citocinas proinflamatorias con satralizumab se considera bajo en cuanto a incidencia y elevación de las citocinas.
SOBREDOSIS: No se han descrito sobredosis en pacientes con NMO o TENMO. En un estudio de fase I se administró una dosis única de hasta 240 mg de ENSPRYNG por vía subcutánea a voluntarios adultos sanos, sin que se observasen acontecimientos adversos graves o intensos durante el estudio.
En caso de sobredosis, se vigilará estrechamente al paciente, se le administrará tratamiento sintomático y se instaurarán las medidas de apoyo que sean necesarias.
PRESENTACIÓN: Caja por una jeringa prellenada de 120 mg (Reg. San. No. INVIMA 2022MBT-0000052)
Condición de venta: Venta bajo fórmula médica.
Mayor información:
PRODUCTOS ROCHE, S. A.
Bogotá, Colombia
CDS 1.0 - Jul 2019


