
FRAXIPARINE
NADROPARINA
Solución inyectable
1 Envase(s), 2 Jeringas prellenadas, 0,3 mL
1 Envase(s), 10 Jeringas prellenadas, 0,3 mL
1 Envase(s), 2 Jeringas prellenadas, 0.4 mL
1 Envase(s), 10 Jeringas prellenadas, 0.4 mL
1 Envase(s), 2 Jeringas prellenadas, 0.6 mL
1 Envase(s), 10 Jeringas prellenadas, 0.6 mL
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN:
Composición cualitativa y cuantitativa:
1 ml de FRAXIPARINE solución inyectable contiene:
Anti-Xa de nadroparina cálcica 9,500 unidades internacionales (UI)
Jeringas prellenadas:
0.3 ml de solución equivalente a 2,850 unidades
internacionales de anti-Xa
0.4 ml de solución equivalente a 3,800 unidades
internacionales de anti-Xa.
Jeringas prellenadas graduadas:
0.6 ml de solución equivalente a 5,700 unidades
internacionales de anti-Xa Para consultar la lista
completa de excipientes, ver sección Lista de excipientes
Forma farmacéutica:
Solución inyectable en jeringa prellenada. La solución es estéril y clara, pH 5.0 a 7.5.
INDICACIONES TERAPÉUTICAS:
Datos clínicos:
• Profilaxia de trastornos tromboembólicos; por ejemplo:
- Los asociados con intervención quirúrgica general u ortopédica
- Pacientes bajo tratamiento médico confinados a cama y con riesgo incrementado de tromboembolismo venoso.
- Aquellos en pacientes de alto riesgo (insuficiencia respiratoria y/o infección respiratoria y/o insuficiencia cardiaca), inmovilizados debido a enfermedad aguda, u hospitalizados en unidad de cuidados intensivos.
• Tratamiento de trastornos tromboembólicos.
• Prevención de coagulación durante hemodiálisis.
• Tratamiento de angina de pecho inestable e infarto del miocardio sin ondas Q.
PROPIEDADES FARMACÉUTICAS:
Propiedades farmacológicas:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Agentes antitrombóticos–Grupo de las heparinas.
Código ATC: B01AB06
Mecanismo de acción: Nadroparina es una heparina de bajo peso molecular fabricada por despolimerización de heparina estándar. Es un glucosaminoglicano con peso molecular medio de aproximadamente 4300 daltons.
Nadroparina presenta alta afinidad de enlace con la proteína plasmática antitrombina III (ATIII). Dicho enlace conduce a inhibición acelerada de factor Xa, la cual contribuye al elevado potencial antitrombótico de nadroparina.
Otros mecanismos que contribuyen a la actividad antitrombótica de nadroparina incluyen estimulación del inhibidor de la vía de factor tisular (TFP1), activación de fibrinólisis mediante liberación directa de activador plasminógeno tisular en células endoteliales, y modificación de parámetros hemorrágicos (reducción de la viscosidad sanguínea e incremento de la fluidez plaquetaria y de la membrana de granulocito).
Efectos farmacodinámicos: Nadroparina tiene una proporción alta de actividad anti-Xa respecto a anti-IIa. Ejerce acción antitrombótica tanto inmediata como prolongada.
En comparación con heparina no fraccionada, nadroparina ejerce menor efecto sobre el funcionamiento de trombocito y su agregación y sólo un leve efecto sobre la hemostasia primaria.
Propiedades farmacocinéticas: Las propiedades farmacocinéticas de nadroparina se han evaluado basándose en su actividad biológica, es decir, medición de actividad anti-factor Xa.
Absorción: Tras la administración subcutánea, se alcanza la actividad anti-Xa máxima (Cmáx) tras aproximadamente 3 a 5 horas (Tmáx).
La biodisponibilidad es casi completa (alrededor de 88%).
Tras inyección IV, se alcanza el nivel máximo plasmático de anti-Xa en menos de 10 minutos, y la vida media es alrededor de 2 horas.
Eliminación: La vida media de eliminación tras inyección subcutánea es aproximadamente 3.5 horas. Sin embargo, la actividad de anti-Xa es detectable por lo menos 18 horas tras la inyección de 19000 UI de anti-Xa.
Poblaciones de pacientes especiales:
Adultos mayores: El funcionamiento renal generalmente disminuye con la edad, de modo que la eliminación es más lenta en ancianos (ver Farmacocinética: Insuficiencia renal). Es preciso considerar la posibilidad de insuficiencia renal en este grupo de edad y, en consecuencia, el ajuste de dosificación (ver Posología y forma de administración, Advertencias especiales y precauciones de empleo).
Insuficiencia renal: En un estudio clínico, donde se investigó la farmacocinética de nadroparina administrada por vía intravenosa a pacientes con grados diversos de insuficiencia renal, se detectó una correlación entre depuración de nadroparina y depuración de creatinina. En pacientes con afección renal moderada (depuración de creatinina de 36-43 ml/min), tanto el ABC medio como la vida media se aumentaron en 52 y 39%, respectivamente, en comparación con voluntarios sanos.
En estos pacientes, la depuración plasmática media de nadroparina se redujo a 63% de lo normal. Se observó amplia variabilidad interindividual en el estudio. En sujetos con insuficiencia renal severa (depuración de creatinina de 10-20 ml/min), tanto el ABC medio como la vida media aumentaron 95 y 112%, respectivamente, en comparación con voluntarios sanos. En pacientes con insuficiencia renal severa, la depuración plasmática disminuyó 50%, en comparación con la observada en pacientes con una función renal normal. En pacientes con insuficiencia renal severa (depuración de creatinina de 3-6 ml/min) sujetos a hemodiálisis, tanto el ABC como la vida media se incrementaron en 62 y 65%, respectivamente, en comparación con voluntarios sanos. La depuración del plasma en pacientes con insuficiencia renal severa sujetos a hemodiálisis disminuyó al 67% de lo observado en pacientes con función renal normal (ver Posología y forma de administración, Advertencias especiales y Precauciones de empleo).
Datos farmacéuticos:
Lista de excipientes: Solución de hidróxido de calcio o ácido clorhídrico diluido para ajuste de pH (5 a 7.5). Agua inyectable.
Incompatibilidades: No mezclar con otros productos.
Periodo de validez: 36 meses.
CONTRAINDICACIONES:
Nadroparina está contraindicada en casos de:
- Hipersensibilidad a la sustancia activa nadroparina o cualquiera de los excipientes listados en la sección Lista de excipientes.
- Antecedentes de trombocitopenia bajo tratamiento con nadroparina y con otras heparinas
- Hemorragia activa o aumento del riesgo de hemorragia, en relación con trastornos hemostáticos, excepto en caso de coagulación intravascular diseminada no inducida por heparina.
- Lesión orgánica con probabilidades de hemorragia (como ulcera péptica activa)
- Accidente cerebrovascular hemorrágico
- Endocarditis infecciosa aguda.
- Afección renal severa (depuración de creatinina inferior a 30 ml/min) en pacientes que reciban tratamiento de trastornos tromboembólicos, angina de pecho inestable e infarto del miocardio sin ondas Q.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Fertilidad, embarazo y lactancia:
Fertilidad: No se tienen estudios clínicos sobre el efecto de nadroparina en la fertilidad.
Embarazo: Los estudios en animales no han demostrado ningún efecto teratógeno o fetotóxico. Sin embargo, sólo se cuenta con datos clínicos limitados respecto al paso transplacentario de nadroparina en embarazadas. Por lo tanto, no se recomienda el uso de nadroparina durante el embarazo, a menos que los beneficios terapéuticos compensen los posibles riesgos.
Lactancia: Las informaciones disponibles sobre la excreción de la nadroparina en la leche materna son limitadas. Por lo tanto, el uso de nadroparina durante la lactancia no es recomendado.
EFECTO EN LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS:
Efectos sobre la capacidad para conducir y utilizar máquinas: No se tienen datos sobre el efecto de nadroparina en el desempeño de manejo o la capacidad para operar maquinaria.
REACCIONES ADVERSAS: Los efectos indeseables pueden variar en su incidencia dependiendo de la dosis recibida y también cuando se administran en combinación con otros agentes terapéuticos.
A continuación, se incluyen las reacciones adversas por sistema de clasificación de órganos y frecuencia.
La siguiente convención se empleó para clasificar las reacciones adversas en términos de frecuencia:
Muy común (≥ 1/10)
Común (≥ 1/100 a < 1/10)
No común (≥ 1/1,000 a < 1/100)
Rara (≥ 1/10,000 a < 1/1,000)
Muy rara (< 1/10,000)
Frecuencia desconocida (no puede estimarse a partir de los datos disponibles).
Resumen tabulado de reacciones adversas:
|
Sistema de clasificación de órganos |
Frecuencia |
Reacción adversa |
|
Trastornos sanguíneos y del sistema linfático |
Muy común |
Hemorragia1 |
|
Rara |
Trombocitopenia, trombocitopenia inducida por heparinas (ver Advertencias especiales y Precauciones de empleo), Trombocitosis. |
|
|
Muy rara |
Eosinofilia, reversible tras la suspensión del tratamiento |
|
|
Trastornos del sistema inmune |
Muy rara |
Reacciones de hipersensibilidad (incluyendo angioedemas y reacciones cutáneas), reacción anafilactoide. |
|
Trastornos del sistema nervioso |
Desconocida |
Dolor de cabeza Migraña |
|
Trastornos del metabolismo y la nutrición |
Muy rara |
Hiperpotasemia reversible relacionada con supresión de aldosterona inducida por heparina, en especial en pacientes en riesgo (ver Advertencias especiales y Precauciones de empleo). |
|
Trastornos mamarios y del sistema reproductivo |
Muy rara |
Priapismo |
|
Trastornos de la piel y el tejido subcutáneo |
Rara |
Exantema, urticaria, eritema, prurito |
|
Investigaciones |
Común |
Aumento de las transaminasas, generalmente transitorio. |
|
Trastornos generales y en el sitio de administración |
Muy común |
Hematoma en el sitio de la inyección2. |
|
Común |
Reacción en el sitio de inyección. |
|
|
Rara |
Calcificación en el sitio de inyección3 |
|
|
Muy rara |
Necrosis en el sitio de inyección (ver Advertencias especiales y Precauciones de empleo). |
1 Manifestaciones hemorrágicas en varios sitios, más frecuente en pacientes con otros factores de riesgo (ver Contraindicaciones e Interacciones).
2 En algunos casos, se puede notar la aparición de nódulos firmes, que no indican un enquistamiento de la heparina. Estos nódulos suelen desaparecer al cabo de unos días.
3 La calcificación es más frecuente en pacientes con producto de fosfato de calcio anormal, como en algunos casos de insuficiencia renal crónica.
Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento después de la autorización. Permite un seguimiento continuo de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de farmacovigilancia. Si presenta cualquier tipo de evento adverso con el uso de este producto, consulte a su médico tratante y repórtelo a farmacovigilancia@aspenlatam.com.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacción con otros medicamentos y otras formas de interacción:
Anticoagulantes orales, (gluco-) corticosteroides sistémicos y dextrano: Nadroparina debe administrarse con precaución en pacientes que reciben anticoagulantes orales, (gluco-) corticosteroides sistémicos y dextrano.
La administración de nadroparina en pacientes que cambian a anticoagulantes orales debe continuarse hasta que se alcance un INR (índice internacional normalizado) estable dentro del rango deseado.
Salicilatos, antiinflamatorios no esteroideos e inhibidores de la agregación plaquetaria: No se recomienda el uso simultáneo de ácido acetilsalicílico (u otros salicilatos), antiinflamatorios no esteroideos e inhibidores de la agregación plaquetaria, ya que pueden aumentar el riesgo de hemorragia (ver Sección Advertencias y precauciones especiales de empleo).
Nitroglicerina: La interacción de la heparina con la nitroglicerina intravenosa, que puede reducir el efecto de la heparina, no puede excluirse para la nadroparina. Los medicamentos que aumentan el nivel de potasio en suero sólo deben usarse bajo un control médico especialmente cuidadoso durante el uso simultáneo de nadroparina.
HALLAZGOS DE LABORATORIO CLÍNICO:
Datos preclínicos sobre seguridad: Los datos preclínicos no revelaron riesgo especial en humanos basándose en estudios convencionales de seguridad farmacológica, toxicidad de dosis repetida, genotoxicidad, potencial mutagénico y toxicología reproductiva.
PRECAUCIONES Y ADVERTENCIAS:
Advertencias y precauciones especiales de empleo:
Reactividad cruzada: La reactividad cruzada entre heparinas y heparinas de bajo peso molecular (HBPM) está bien documentada. Se han notificado reacciones de hipersensibilidad retardada en pacientes que presentaban reactividad cruzada entre heparinas no fraccionadas y HBPM.
Antes de iniciar la terapia con HBPM, se debe realizar una evaluación cuidadosa de las reacciones previas de hipersensibilidad a la heparina no fraccionada.
Trombocitopenia inducida por heparinas: Debido a la posibilidad de trombocitopenia inducida por heparina, debe realizarse conteo de plaquetas durante todo el curso de tratamiento con nadroparina.
Se han reportado ocasionalmente casos raros de trombocitopenia ocasionalmente severa, la cual podría asociarse con trombosis arterial o venosa. Dicho diagnóstico debe considerarse en las siguientes situaciones:
- Trombocitopenia.
Cualquier reducción significativa del nivel de plaquetas (30 a 50% comparada al valor basal).
- Empeoramiento de trombosis inicial mientras esté bajo terapia.
- Trombosis ocurrida durante el tratamiento.
- Coagulación intravascular diseminada.
En este caso, debe descontinuarse el tratamiento con nadroparina.
Estos efectos probablemente sean de naturaleza inmuno-alérgica, y en el caso de ser el primer tratamiento, se reportan principalmente entre el quinto y el veintiún día de terapia, pero pueden ocurrir en etapa más temprana si hay historia de trombocitopenia inducida por heparina.
En caso de antecedentes de trombocitopenia bajo tratamiento con heparina (ya sea heparina estándar o de bajo peso molecular), puede considerarse en caso necesario, tratamiento con nadroparina. En este tipo de casos, será preciso realizar monitoreo clínico cuidadoso y valoración de conteo de plaquetas por lo menos una vez al día. Si ocurre trombocitopenia, el tratamiento debe descontinuarse de inmediato.
Cuando ocurra trombocitopenia con heparina (ya sea heparina estándar o de bajo peso molecular), debe considerarse la posibilidad de sustitución con diferente tipo de antitrombótico. Si no se dispone del mismo, entonces podrá considerarse la sustitución por otra heparina de bajo peso molecular, si es necesaria la administración de heparina. En estos casos se realizará monitoreo de conteo de plaquetas por lo menos una vez al día y se descontinuará el tratamiento tan pronto sea posible, pues se han descrito casos de trombocitopenia inicial que continúa tras la sustitución (ver Contraindicaciones).
Las pruebas de agregación plaquetaria in vitro son tan sólo de valor limitado en el diagnóstico de trombocitopenia inducida por heparina.
Debe tenerse precaución al administrar nadroparina en las siguientes situaciones, ya que podrían asociarse con aumento del riesgo de hemorragia:
- Insuficiencia hepática.
- Hipertensión arterial severa.
- Historia de úlcera péptica u otra lesión orgánica con probabilidad de hemorragia.
- Trastorno vascular de la corio-retina.
- Durante periodo postoperatorio tras intervención quirúrgica cerebral de la médula espinal u ocular.
- Tratamiento simultáneo con anticoagulantes orales.
Hiperpotasemia: La heparina puede suprimir la secreción suprarrenal de aldosterona conduciendo a hiperpotasemia, en particular en pacientes con aumento de potasio en plasma, o con riesgo de aumento de niveles plasmáticos de potasio como pacientes con diabetes mellitus, insuficiencia renal crónica, acidosis metabólica preexistente y a quienes toman fármacos que puedan provocar hiperpotasemia (p. ej., inhibidores de la enzima convertidora de angiotensina (ACE), fármacos antiinflamatorios no esteroides (AINE).
Aparentemente, el riesgo de hiperpotasemia aumenta con la duración de la terapia, pero suele ser reversible.
En los pacientes en riesgo debe monitorearse el potasio en plasma.
Anestesia espinal/epidural/punción espinal lumbar y fármacos concomitantes: El riesgo de hematomas espinales/epidurales se incrementa cuando hay catéter epidural fijo o por el uso concomitante de otros fármacos que afectan la hemostasia como AINE, inhibidores de plaquetas u otros anticoagulantes. Aparentemente, este riesgo también se incrementa en caso de punción epidural o espinal traumático o repetido.
Por lo tanto, debe decidirse la prescripción concomitante de bloqueo neuroaxial y de una terapia anticoagulante tras evaluación cuidadosa de la proporción de beneficio/riesgo individual en los siguientes casos:
• En pacientes que ya reciban tratamiento con anticoagulantes, se evaluará cuidadosamente los beneficios de bloqueo neuroaxial contra los riesgos.
• En pacientes que se planea someter a intervención quirúrgica programada con bloqueo neuroaxial, se evaluarán cuidadosamente los beneficios de la terapia con anticoagulante contra los riesgos.
En el caso de pacientes con punción espinal lumbar, anestesia espinal o anestesia epidural, deben transcurrir por lo menos 12 horas entre la inyección de nadroparina a dosis profilácticas, o 24 horas a dosis terapéuticas y la inserción o eliminación del catéter o aguja espinal/epidural. En pacientes con insuficiencia renal, se podrían contemplar intervalos mayores.
Se monitoreará con frecuencia a los pacientes para signos y síntomas de insuficiencia neurológica. Si se observa compromiso neurológico, será necesario tratamiento de urgencia.
Salicilatos, antiinflamatorios no esteroides y fármacos antiplaquetarios: En la profilaxis o tratamiento de trastornos de tromboembolia venosa, y en la prevención de coagulación durante hemodiálisis, no se recomienda el uso concomitante de aspirina, otros salicilatos, AINE y agentes antiplaquetarios, ya que podrían aumentar el riesgo de hemorragia. Cuando no se puede evitar ese tipo de combinación se debe realizar monitoreo clínico y biológico cuidadoso.
En estudios clínicos para tratar angina de pecho inestable e infarto del miocardio sin ondas Q, nadroparina se administró combinada con hasta 325 mg de aspirina al día (ver Posología y forma de administración).
Poblaciones especiales:
Población pediátrica: No se dispone de experiencia clínica sobre el uso de nadroparina en niños. Por esta razón, no se recomienda el uso de nadroparina en niños hasta que se disponga de datos adicionales.
Adultos mayores: Se recomienda evaluar el funcionamiento renal antes de iniciar el tratamiento (ver Contraindicaciones).
Insuficiencia renal: Se sabe que la nadroparina es excretada principalmente por los riñones, lo cual produce un aumento en la exposición a la nadroparina en pacientes con insuficiencia renal (ver Farmacocinética-Insuficiencia renal). Los pacientes con insuficiencia renal se encuentran en mayor riesgo de presentar hemorragias, por lo cual deberán ser tratados con precaución.
La decisión sobre si una reducción de la dosis es adecuada para pacientes con un aclaramiento de creatinina de 30 a 50 ml/min debe basarse en la evaluación del médico del riesgo de hemorragia de un paciente individual frente al riesgo de tromboembolismo (ver Posología y forma de administración).
Necrosis cutánea: En muy raras ocasiones se han reportado casos de necrosis cutánea. Es precedida por púrpura o manchas eritematosas infiltradas o dolorosas, con o sin signos generales. En esos casos, se deberá suspender inmediatamente el tratamiento.
Jeringas prellenadas graduadas:
Alergia al látex: El capuchón de la aguja de la jeringa prellenada puede contener caucho de látex natural que tiene el potencial de causar reacciones alérgicas severas en individuos sensibles al látex.
Precauciones especiales de eliminación y otras manipulaciones:
Información sobre la manipulación: Nadroparina se examinará visualmente para detectar partículas de materia y decoloración antes de su uso. Utilizar únicamente soluciones inyectables cuyo aspecto no haya cambiado. Para un solo uso. Desechar cualquier solución residual no utilizada.
Cualquier medicamento no utilizado o material de desecho debe eliminarse de acuerdo con los requisitos locales.
Instrucciones paso a paso:
Partes de la jeringa prellenada de FRAXIPARINE:
1 Tapa protectora de la aguja.
2 Émbolo.
3 Soporte para los dedos.
4 Protección de seguridad.

Información sobre el uso:
1. Lave bien sus manos con agua y jabón y séquelas con una toalla.
2. Retire la jeringa de la caja y compruebe que:
La fecha de vencimiento de la caja, y sobre la jeringa prellenada que la jeringa no haya sido abierta o esté dañada.
3. Siéntese o recuéstese en una posición cómoda. Escoja un lugar en el área abdominal inferior (barriga), al menos 5 cm por debajo del ombligo (Figura A).
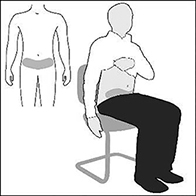
Figura A
Al administrar cada inyección, alterne el lado izquierdo y derecho del área abdominal baja. Esto ayudará a reducir el malestar en el sitio de inyección. Si la inyección en el área abdominal baja no es posible, pida consejo a su enfermera o médico (Figura A).
4. Limpie el área de inyección con un algodón con alcohol.
5. Retire la tapa que protege la aguja, primero girándola y luego empujándola en una línea recta lejos del cuerpo de la jeringa (Figura B).
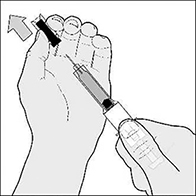
Figura B
Deseche la tapa protectora de la aguja: Si el volumen contenido en la jeringa es mayor a lo que usted necesita, deberá retirar el excedente antes de inyectarse.
• Sostenga la jeringa con la aguja apuntando verticalmente hacia abajo.
• Presione suavemente el émbolo de la jeringa hacia abajo hasta que la burbuja se encuentre en la línea marcada con el volumen que su médico le haya prescrito.
• Deje gotear en algún papel el líquido que salga de la aguja y deséchelo.
• La jeringa ya está lista para usarse.
Nota importante:
• No toque la aguja, ni permita que ésta entre en contacto con alguna superficie, antes de aplicar la inyección
La presencia de una pequeña burbuja de aire en la jeringa es normal. No intente retirar esta burbuja de aire antes de aplicar la inyección – Podría perder parte del medicamento si lo hace.
6. Pellizque suavemente la piel previamente limpia, con la finalidad de hacer un pliegue. Sostenga el pliegue entre sus dedos pulgar e índice durante toda la aplicación de la inyección (Figura C).
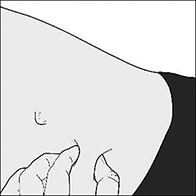
Figura C
7. Sostenga firmemente la jeringa del soporte para los dedos. Inserte verticalmente (a un ángulo de 90°) toda la aguja en el pliegue de piel (Figura D).
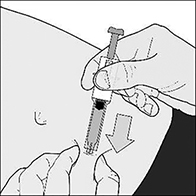
Figura D
8. Inyecte TODO el contenido de la jeringa presionando el émbolo hasta el fondo (Figura E). A continuación, retire suavemente la aguja de la piel.
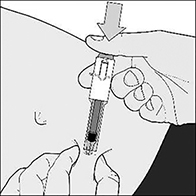
Figura E
9. Después de aplicar la inyección, sostenga la jeringa con una mano, sujetando la protección de seguridad, luego utilice la otra mano para jalar firmemente el soporte para los dedos. Esto desbloquea la protección. Deslice la protección hacia arriba del cuerpo de la jeringa, hasta que quede bloqueada en su posición sobre la aguja (Figura F).
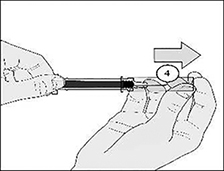
Figura F
DOSIS Y VÍA DE ADMINISTRACIÓN:
Posología y forma de administración:
General: Debe prestarse particular atención a las instrucciones específicas de dosificación de productos patentados como Heparina de Bajo Peso Molecular, ya que se emplean distintas unidades de medición (unidades o mg) para expresar la dosificación. Por lo tanto, nadroparina no debe emplearse de manera intercambiable con otras heparinas de bajo peso molecular durante tratamiento continuo. Además, se debe tener cuidado de emplear la formulación correcta de nadroparina, ya sea de concentración simple o doble, ya que esto afectará el régimen de dosificación. Las jeringas graduadas se usan cuando es necesario el ajuste de dosis por peso del cuerpo.
El conteo de plaquetas debe monitorearse durante todo el tratamiento con nadroparina (ver Advertencias y precauciones especiales de empleo).
Se deben seguir las recomendaciones específicas respecto a los tiempos de dosificación con nadroparina alrededor de la administración de anestesia espinal/epidural o de la punción lumbar (ver Advertencias y precauciones especiales de empleo).
Posología:
Adultos:
Profilaxia de trastornos tromboembólicos:
Cirugía general: La dosis recomendada de Fraxiparine es 0.3 ml (2,850 UI de anti-Xa) administrados por vía subcutánea de 2 a 4 horas antes de la intervención quirúrgica, y después una vez al día en días subsecuentes. El tratamiento debe continuarse cuando menos por siete días y durante todo el periodo de riesgo, hasta que el paciente sea ambulatorio.
Cirugía ortopédica: FRAXIPARINE se administra por vía subcutánea y se ajusta a la dosis según el peso del cuerpo tomando en cuenta la siguiente tabla. Ésta se basa en una dosis meta de 38 UI de anti-Xa por kg de peso del cuerpo y se incrementa en 50% en el cuarto día postoperatorio. La dosis inicial se administra 12 horas antes de la intervención quirúrgica, administrando una segunda dosis 12 horas después de finalizada la cirugía. Posteriormente, el tratamiento se continúa una vez al día durante todo el periodo de riesgo y hasta que el paciente sea ambulatorio. El periodo mínimo del tratamiento es 10 días.
|
Peso del cuerpo (kg) |
12 horas antes y después de la intervención quirúrgica y posteriormente una vez al día hasta el tercer día postoperatorio |
Desde el cuarto día del postoperatorio en adelante |
||
|
Volumen inyectado (ml) |
UI de anti-Xa |
Volumen inyectado (ml) |
UI de anti-Xa |
|
|
< 50 50-69 ≥ 70 |
0.2 0.3 0.4 |
1,900 2,850 3,800 |
0.3 0.4 0.6 |
2,850 3,800 5,700 |
- Pacientes de alto riesgo (insuficiencia respiratoria y/o infección respiratoria y/o insuficiencia cardiaca), inmovilizados debido a enfermedad aguda, u hospitalizados en una unidad de cuidados intensivos.
FRAXIPARINE se administra por vía subcutánea una vez al día. La dosis debe ajustarse según el peso del cuerpo y de acuerdo con la siguiente tabla. El tratamiento se continuará durante todo el periodo de riesgo de tromboembolia.
|
Peso del cuerpo (kg) |
Una vez al día |
|
|
Volumen inyectado (ml) |
UI de anti-Xa |
|
|
≤ 70 |
0.4 |
3,800 |
|
> 70 |
0.6 |
5,700 |
En adultos mayores, puede ser apropiada una reducción de dosis a 0,3 ml (2.850 UI anti Xa)
Tratamiento de trastornos tromboembólicos: En el tratamiento de trastornos tromboembólicos, la terapia con anticoagulantes orales debe iniciarse tan pronto sea posible, a menos que esté contraindicada. El tratamiento con FRAXIPARINE no debe detenerse antes de que se alcance la Proporción Internacional Normalizada (INR, por su sigla en inglés).
Se recomienda administrar FRAXIPARINE por vía subcutánea dos veces al día (cada 12 horas) por un periodo usual de 10 días. Posteriormente, la dosis deberá ajustarse según el peso del cuerpo de acuerdo con la siguiente tabla, la cual se basa en una dosis meta de 86 UI de anti-Xa por kg de peso del cuerpo.
|
Peso del cuerpo (kg) |
Dos veces al día por el periodo usual de 10 días |
|
|
Volumen inyectado (ml) |
UI de anti-Xa |
|
|
< 50 |
0.4 |
3,800 |
|
50-59 |
0.5 |
4,750 |
|
60-69 |
0.6 |
5,700 |
|
70-79 |
0.7 |
6,650 |
|
80-89 |
0.8 |
7,600 |
|
≥ 90 |
0.9 |
8,550 |
Prevención de coagulación durante la hemodiálisis: Para prevenir la coagulación en el curso de la hemodiálisis, la dosis de FRAXIPARINE debe optimizarse para cada paciente individual, tomando en cuenta también las condiciones técnicas de la diálisis.
La nadroparina es administrada como una dosis única en la línea arterial al iniciarse cada sesión. En pacientes sin aumento de riesgo de hemorragia, se sugieren las siguientes dosis iniciales según el peso del cuerpo, las cuales suelen bastar para una sesión de cuatro horas:
|
Peso del cuerpo (kg) |
Inyección a la línea arterial al iniciarse la diálisis |
|
|
Volumen inyectado (ml) |
UI de anti-Xa |
|
|
< 50 |
0.3 |
2,850 |
|
50-69 |
0.4 |
3,800 |
|
≥ 70 |
0.6 |
5,700 |
Las dosis deben reducirse a la mitad en pacientes con riesgo aumentado de hemorragia.
Puede administrarse una dosis adicional más pequeña en el curso de la diálisis en sesiones que duren más de cuatro horas. La dosis en sesiones subsecuentes de diálisis deberá ajustarse según se requiera de acuerdo con el efecto observado.
Los pacientes deben ser monitoreados cuidadosamente durante cada sesión de diálisis buscando signos de sangrado o trombos en el circuito de diálisis.
Tratamiento de angina inestable e infarto del miocardio con onda No-Q: Se recomienda que la nadroparina sea administrada de forma subcutánea dos veces al día (cada 12 horas).
La duración usual del tratamiento es de seis días. En los estudios clínicos en pacientes con angina inestable e infarto del miocardio con onda no-Q, la nadroparina fue administrada en combinación con hasta 325 mg de aspirina al día.
La dosis inicial es administrada como un bolo de inyección intravenosa (IV) y subsecuentemente, por dosis en inyecciones subcutáneas. La dosis debe ser ajustada para peso corporal de acuerdo con la siguiente tabla, la cual es basada para una dosis meta de 86 UI anti-Xa por kg de peso corporal.
|
Peso del cuerpo (kg) |
Inyección intravenosa rápida inicial |
Inyección subcutánea (cada 12 horas) |
UI de anti-Xa |
|
< 50 |
0.4 ml |
0.4 ml |
3,800 |
|
50-59 |
0.5 ml |
0.5 ml |
4,750 |
|
60-69 |
0.6 ml |
0.6 ml |
5,700 |
|
70-79 |
0.7 ml |
0.7 ml |
6,650 |
|
80-89 |
0.8 ml |
0.8 ml |
7,600 |
|
90-99 |
0.9 ml |
0.9 ml |
8,550 |
|
≥ 100 |
1.0 ml |
1.0 ml |
9,500 |
Población pediátrica:
Nadroparina no se recomienda en niños y adolescentes, ya que se cuenta con datos insuficientes de seguridad y eficacia para establecer la dosificación en pacientes de menos de 18 años.
Adultos mayores:
- Profilaxis de desórdenes tromboembólicos en cirugía, prevención de la coagulación en hemodiálisis y tratamiento de Angina Inestable e Infarto de Miocardio sin Onda Q y TEV No se requiere ajuste de dosis en ancianos, a menos que el funcionamiento renal se encuentre alterado. Se recomienda valorar el funcionamiento renal antes de iniciar el tratamiento (ver a continuación Afecciones renales y Farmacocinética).
- Profilaxis de desórdenes tromboembólicos en pacientes de alto riesgo médico (falla respiratoria y/o infección respiratoria y/o falla cardiaca), inmovilizados debido a enfermedades agudas u hospitalizadas en una unidad de cuidado intensivo.
En pacientes ancianos, una reducción de la dosis a 0.3 ml (2,850 UI anti-Xa) puede ser apropiada.
Insuficiencia renal:
- Profilaxia de trastornos tromboembólicos:
• No se requiere reducción de la dosis en pacientes con afección renal leve (depuración de creatinina mayor o igual a 50 ml/min).
• La afección renal moderada y severa se encuentra asociada con un aumento en la exposición a la nadroparina. Estos pacientes se encuentran en mayor riesgo de presentar tromboembolia y hemorragias.
• Si el médico prescriptor considera adecuado reducir la dosificación, tomando en consideración los factores individuales de riesgo de tromboembolia y hemorragia en pacientes con afección renal moderada (depuración de creatinina mayor o igual a 30 ml/min, e inferior a 50 ml/min), la dosis deberá reducirse en 25 a 33% (ver Advertencias y precauciones y Farmacocinéticas).
• La dosis debe reducirse en 25 a 33% en pacientes con afección renal severa (depuración de creatinina inferior a 30 ml/min) (ver Advertencias y precauciones y Farmacocinéticas).
- Tratamiento de trastornos tromboembólicos, angina de pecho inestable e infarto del miocardio sin ondas-Q:
• No se requiere reducir la dosificación en pacientes con afección renal leve (depuración de creatinina mayor o igual a 50 ml/min).
La afección renal moderada y severa se encuentra asociada con un aumento en la exposición a la nadroparina. Estos pacientes se encuentran en mayor riesgo de presentar tromboembolia y hemorragias.
• Si el médico prescriptor considera adecuado reducir la dosificación, tomando en consideración los factores individuales de riesgo de tromboembolia y hemorragia en pacientes con afección renal moderada (depuración de creatinina mayor o igual a 30 ml/min, e inferior a 50 ml/min), la dosis deberá reducirse en 25 a 33% (ver Advertencias especiales y precauciones de empleo y Farmacocinéticas).
Nadroparina está contraindicada en pacientes con afecciones renales severas (depuración de creatinina menor a 30 ml/min (ver Contraindicaciones).
Insuficiencia hepática: No se han realizado estudios en pacientes con afección hepática.
Forma de administración: La nadroparina no está destinada a inyección intramuscular.
El sitio común para la inyección subcutánea es el lado derecho o izquierdo de la pared abdominal, pero también puede emplearse el muslo como alternativa. Para evitar pérdida de la solución al usar jeringas prellenadas, la burbuja de aire no debe expulsarse de la jeringa antes de la inyección. La aguja debe insertarse perpendicularmente a un pliegue cutáneo detenido entre el pulgar y el índice de manera suave pero firme hasta que se haya completado la inyección. No se debe frotar el sitio de inyección.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Sobredosis:
Síntomas y signos: La hemorragia es el principal signo clínico de sobredosis subcutánea o intravenosa. Deberá medirse el conteo de plaquetas y otros parámetros de coagulación. La hemorragia menor rara vez requiere de terapia específica; suele bastar con reducir o retrasar las dosis subsecuentes de nadroparina.
Tratamiento: Debe considerarse el uso de sulfato de protamina únicamente en casos serios. Ésta neutraliza principalmente el efecto anticoagulante de nadroparina, pero aún quedará cierta actividad de anti-Xa.
0.6 ml de sulfato de protamina neutraliza aproximadamente 950 UI de anti-Xa de nadroparina. La cantidad de protamina por inyectar se calculará tomando en cuenta el tiempo transcurrido desde la inyección de heparina y quizá sea adecuado reducir la dosis de protamina.
PRESENTACIÓN:
Naturaleza y contenido del envase: Jeringa prellenada compuesta por un cilindro de vidrio tipo I.
Envase con 2 y 10 jeringas prellenadas con 0,3 ml y 0.4ml de solución inyectable cada una.
Envase con 2 y 10 jeringas prellenadas graduadas con 0.6 ml de solución inyectable cada una.
Es posible que no se comercialicen todos los tamaños de envase.
ASPEN LABS
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Precauciones especiales de conservación:
No se congele. No se refrigere, pues la inyección fría puede resultar dolorosa. No se almacene a más de 30 °C.


