

JARDIANCE DUO®
EMPAGLIFLOZINA, METFORMINA
Tabletas recubiertas
Tabletas recubiertas, 5/850 mg/mg
Tabletas recubiertas, 5/1000 mg/mg
Tabletas recubiertas, 12.5/1000 mg/mg
Tabletas recubiertas, 12.5/850 mg/mg
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN: Un COMPRIMIDO Recubierto contiene: D-Glucitol,1,5-anhidro-1-C-[4-cloro-3-[[4-[[(3S)-tetrahidro-3-furanil]oxi]fenil]metil]fenil]-, (1S) (= empagliflozina) 5 o 12,5 mg y clorhidrato de N,N-dimetil imidodicarbonimídico diamida (= clorhidrato de metformina) 500 mg, 850 mg o 1000 mg
INDICACIONES: JARDIANCE DUO® está indicado como tratamiento complementario a un régimen de dieta y ejercicio físico para mejorar el control glucémico en pacientes adultos con diabetes mellitus tipo 2:
1. En los que no se logra un control adecuado con metformina
2. En los que no se logra un control adecuado con metformina en combinación con otros hipoglucemiantes, incluida la insulina
3. Que ya están recibiendo tratamiento con empagliflozina y metformina coadministradas como comprimidos con cada fármaco por separado.
4. Para reducir el riesgo de muerte cardiovascular en pacientes adultos con diabetes mellitus tipo 2 y enfermedad cardiovascular establecida acompañado de otras medidas que reduzcan el riesgo cardiovascular.
USO EN POBLACIONES ESPECÍFICAS: Fertilidad, embarazo y lactancia: Embarazo: Los datos que existen sobre el uso de JARDIANCE DUO® o de sus componentes individuales en mujeres embarazadas son limitados. Los estudios preclínicos realizados con empagliflozina sola no indican efectos nocivos directos ni indirectos en lo que se refiere a la toxicidad para la reproducción. Los estudios en animales efectuados con la combinación de empagliflozina y metformina o con metformina sola han indicado toxicidad reproductiva para el caso de las dosis altas de metformina únicamente (véase la sección Toxicología).
Como medida de precaución, se recomienda evitar el uso de JARDIANCE DUO® durante el embarazo a menos que sea claramente necesario.
Lactancia: La metformina se excreta en la leche materna en los seres humanos. No se observó ningún efecto adverso en los neonatos/lactantes alimentados con leche materna. Se desconoce si la empagliflozina se excreta en la leche humana.
Los datos preclínicos disponibles obtenidos en animales han indicado la excreción de empagliflozina en la leche. No se puede excluir la posibilidad de un riesgo para los neonatos/lactantes humanos. Se recomienda interrumpir la lactancia durante el tratamiento con JARDIANCE DUO®.
Fertilidad: No se han llevado a cabo estudios sobre el efecto de JARDIANCE DUO® o sus componentes individuales sobre la fertilidad en los seres humanos.
Los estudios preclínicos en animales efectuados con los componentes individuales no indican efectos nocivos directos ni indirectos en lo que a la fertilidad se refiere.
Capacidad para conducir vehículos y operar maquinaria: No se han realizado estudios en torno a los efectos de este producto sobre la capacidad para conducir vehículos y operar maquinaria.
PROPIEDADES FARMACOLÓGICAS: Grupo farmacoterapéutico: Combinaciones de fármacos hipoglucemiantes orales, código ATC: A10BD20
Modo de acción: La empagliflozina es un inhibidor competitivo, selectivo, reversible y altamente potente del SGLT-2, con un valor de IC50 de 1,3 nM. Tiene una selectividad 5000 veces mayor frente al SGLT-1 humano (IC50 de 6278 nM), responsable de la absorción de glucosa en los intestinos. También se demostró una elevada selectividad para otros transportadores de glucosa (GLUT) responsables de la homeostasis de la glucosa en los diferentes tejidos.
La expresión de SGLT-2 en los riñones es elevada, mientras que en otros tejidos este transportador está ausente o su expresión es muy baja. Este transportador es el principal responsable de la reabsorción de glucosa del filtrado glomerular hacia el torrente sanguíneo. En los pacientes con diabetes mellitus tipo 2 (DMT2) e hiperglucemia, la cantidad de glucosa que se filtra y se reabsorbe es mayor.
La empagliflozina mejora el control glucémico en los pacientes con DMT2 mediante la reducción de reabsorción de glucosa renal. La cantidad de glucosa eliminada por el riñón a través de este mecanismo glucurético varía en función de la concentración de glucosa en sangre y la TFGe. Mediante la inhibición del SGLT-2 en los pacientes con DMT2 e hiperglucemia, el exceso de glucosa se excreta a través de la orina.
En los pacientes con DMT2, la excreción de glucosa urinaria se incrementó inmediatamente después de la primera dosis de empagliflozina y se mantiene durante el intervalo de 24 horas entre dosis. El aumento de la excreción de glucosa urinaria se mantuvo al final del período de tratamiento de 4 semanas, con un valor promedio de aproximadamente 78 g/día con dosis de empagliflozina de 25 mg administradas en una toma diaria. Este incremento de la excreción de glucosa urinaria condujo a una reducción inmediata de los niveles de glucosa plasmática en los pacientes con DMT2.
La empagliflozina mejora los niveles plasmáticos de glucosa tanto pre- como posprandiales.
El mecanismo de acción de la empagliflozina es independiente de la función de las células beta y la vía metabólica de insulina, y esto contribuye a que el riesgo de hipoglucemia con este fármaco sea bajo.
Se observó una mejora de los marcadores sustitutos de la función de las células beta, incluyendo el HOMA-β (Homeostasis Model Assessment-B) y el cociente proinsulina-insulina. Además, la excreción de glucosa urinaria desencadena un fenómeno de pérdida calórica, asociado con pérdida de grasa corporal y reducción del peso corporal.
La glucosuria que se observa con la empagliflozina está acompañada de una ligera diuresis que podría contribuir a una reducción moderada y sostenida de la presión arterial.
La metformina es una biguanida con efectos antihiperglucémicos, que reduce los valores de glucosa plasmática tanto basales como posprandiales. Este fármaco no estimula la secreción de insulina y, por ende, no provoca hipoglucemia.
El clorhidrato de metformina puede actuar a través de 3 mecanismos de acción:
1. reducción de la producción de glucosa hepática, mediante la inhibición de la gluconeogénesis y la glucogenólisis;
2. en los músculos, incrementando la sensibilidad a la insulina, lo cual se traduce en una mejor captación y utilización de la glucosa periférica; y
3. retardo de la absorción intestinal de la glucosa.
El clorhidrato de metformina estimula la síntesis de glucógeno intracelular a través de su acción sobre la glucógeno sintasa.
El clorhidrato de metformina aumenta la capacidad de transporte de todos los tipos de GLUT de membrana conocidos hasta la fecha.
En los seres humanos, independientemente de su acción sobre la glucemia, el clorhidrato de metformina ejerce efectos favorables sobre el metabolismo de los lípidos. Esto ha sido demostrado en dosis terapéuticas en estudios clínicos controlados a mediano o largo plazo: el clorhidrato de metformina reduce los niveles de colesterol total, colesterol LDL y triglicéridos.
Estudios clínicos: Un total de 10224 pacientes con diabetes tipo 2 fueron tratados en 9 estudios clínicos doble ciego, comparativos con placebo o con tratamiento activo de un mínimo de 24 semanas de duración, de los cuales 2947 pacientes recibieron empagliflozina 10 mg y 3703 recibieron empagliflozina 25 mg como complemento de un tratamiento de metformina.
El tratamiento con empagliflozina en combinación con metformina con o sin otro régimen de base (pioglitazona, sulfonilurea, inhibidores de la DPP-4 e insulina) condujo a mejoras clínicamente relevantes en los valores de HbA1c, glucosa plasmática en ayunas (GPA), peso corporal y presión arterial sistólica y diastólica. La administración de empagliflozina 25 mg condujo a una mayor proporción de pacientes que alcanzaron el valor objetivo de HbA1c de <7% y a un menor número de pacientes que requirieron rescate glucémico en comparación con empagliflozina 10 mg y placebo. Hubo una mejora clínicamente significativa en los valores de HbA1c en todos los subgrupos de sexo, raza, región geográfica, tiempo desde el diagnóstico de DMT2 e índice de masa corporal (IMC). En los pacientes de 75 años de edad o más, se observaron reducciones numéricamente menores en los valores de HbA1c con el tratamiento de empagliflozina. La presencia de valores más elevados de HbA1c en el nivel basal estuvo asociada con una mayor reducción de la HbA1c. La empagliflozina en combinación con la metformina en pacientes sin tratamiento previo condujo a reducciones clínicamente significativas en HbA1c, GPA, peso corporal y Presión Arterial (PA).
Empagliflozina como complemento del tratamiento con metformina: Se realizó un estudio doble ciego, comparativo con placebo de 24 semanas de duración para evaluar la eficacia y la seguridad de empagliflozina en pacientes que no obtuvieron un beneficio terapéutico suficiente con metformina. El tratamiento con empagliflozina condujo a mejoras estadísticamente significativas en la HbA1c y en el peso corporal, y también a reducciones clínicamente significativas en los valores de GPA y presión arterial en comparación con placebo (Tabla 4).
En la extensión doble ciego, comparativa con placebo de este estudio, las reducciones de la HbA1c (cambio respecto del nivel basal de -0,62% para empagliflozina 10 mg, -0,74% para empagliflozina 25 mg y -0,01% para placebo), peso corporal (cambio respecto del nivel basal de -2,39 kg para empagliflozina 10 mg, -2,65 kg para empagliflozina 25 mg y -0,46 kg para el placebo) y presión arterial (PAS [presión arterial sistólica]: cambio respecto del nivel basal de -5,2 mmHg para empagliflozina 10 mg, -4,5 mmHg para empagliflozina 25 mg y -0,8 mmHg para el placebo; PAD [presión arterial diastólica]: cambio respecto del nivel basal de -2,5 mmHg para empagliflozina 10 mg, -1,9 mmHg para empagliflozina 25 mg y -0,5 mmHg para el placebo) se mantuvieron hasta la Semana 76.
|
Tabla 4. Resultados de un estudio comparativo con placebo de 24 semanas (LOCF)3 de empagliflozina como tratamiento complementario de un régimen de metformina (Grupo Completo de Análisis) |
|||
|
Empagliflozina como complemento del tratamiento con metformina |
Placebo |
Empagliflozina 10 mg |
Empagliflozina 25 mg |
|
N |
207 |
217 |
213 |
|
HbA1c (%) |
|||
|
Nivel basal (media) |
7,90 |
7,94 |
7,86 |
|
Cambio respecto del nivel basal1 |
-0,13 |
-0,70 |
-0,77 |
|
Diferencia respecto del placebo1 (IC 97,5%) |
-0,57* (-0,72, -0,42) |
-0,64* (-0,79, -0,48) |
|
|
N |
184 |
199 |
191 |
|
Pacientes (%) que lograron valores de HbA1c <7% con un valor basal de HbA1c ≥7%2 |
12,5 |
37,7 |
38,7 |
|
N |
207 |
216 |
213 |
|
GPA (mg/dl) [mmol/l]2 |
|||
|
Nivel basal (media) |
156,0 [8,66] |
154,6 [8,58] |
149,4 [8,29] |
|
Cambio respecto del nivel basal1 |
6,4 [0,35] |
-20,0 [-1,11] |
-22,3 [-1,24] |
|
Diferencia respecto del placebo1 (IC 95%) |
-26,4* (-31,3, -21,6) [-1,47* (-1,74, -1,20)] |
-28,7* (-33,6, -23,8) [-1,59* (-1,86, -1,32)] |
|
|
N |
207 |
217 |
213 |
|
Peso corporal (kg) |
|||
|
Nivel basal (media) |
79,73 |
81,59 |
82,21 |
|
Cambio respecto del nivel basal1 |
-0,45 |
-2,08 |
-2,46 |
|
Diferencia respecto del placebo1 (IC 97,5%) |
-1,63* (-2,17, -1,08) |
-2,01* (-2,56, -1,46) |
|
|
N |
207 |
217 |
213 |
|
Pacientes (%) que lograron una pérdida de peso de > 5%2 |
4,8 |
21,2 |
23,0 |
|
N |
207 |
217 |
213 |
|
PAS (mmHg)2 |
|||
|
Nivel basal (media) |
128,6 |
129,6 |
130,0 |
|
Cambio respecto del nivel basal1 |
-0,4 |
-4,5 |
-5,2 |
|
Diferencia respecto del placebo1 (IC 95%) |
-4,1* (-6,2, -2,1) |
-4,8* (-6,9, -2,7) |
|
|
1 Media ajustada para el valor basal y estratificación. 2 No evaluado para la determinación de la significancia estadística; no forma parte del procedimiento analítico secuencial aplicado para los criterios de valoración secundarios. 3 Extrapolación de la última observación (previa al rescate glucémico) (LOCF) * Valor p <0,0001. |
|||
Tratamiento combinado de empagliflozina y metformina en pacientes sin tratamiento previo: Se realizó un estudio de diseño factorial de 24 semanas de duración a fin de evaluar la eficacia y la seguridad de empagliflozina en pacientes sin tratamiento previo. El tratamiento con empagliflozina en combinación con metformina (5 mg y 500 mg; 5 mg y 1000 mg; 12,5 mg y 500 mg, y 12,5 mg y 1000 mg administrados dos veces al día) brindó mejoras estadísticamente significativas en la HbA1c y condujo a reducciones significativamente mayores en los valores de GPA y peso corporal en comparación con los componentes individuales. Una mayor proporción de pacientes con un valor basal de HbA1c ≥7,0% y tratados con empagliflozina en combinación con metformina logró un valor objetivo de HbA1c <7% en comparación con los componentes individuales (Tablas 5 y 6).
|
Tabla 5. Resultados de un estudio de 24 semanas (OC [Casos Observados])2 en el que se comparó la empagliflozina 10 mg en combinación con metformina frente a los componentes individuales |
|||||
|
Empagliflozina 10 mg +metformina 1000 mga |
Empagliflozina 10 mg+ metformina 2000 mga |
Empagliflozina 10 mg (una vez al día) |
Metformina 1000 mga |
Metformina 2000 mga |
|
|
N |
161 |
167 |
169 |
167 |
162 |
|
HbA1c (%) |
|||||
|
Nivel basal (media) |
8,7 |
8,7 |
8,6 |
8,7 |
8,6 |
|
Cambio respecto del nivel basal1 |
-2,0 |
-2,1 |
-1,4 |
-1,2 |
-1,8 |
|
Comparación vs. empagliflozina (IC 95%)1 |
-0,6* (-0,9, -0,4)b |
-0,7* (-1,0, -0,5)b |
|||
|
Comparación vs. metformina (IC 95%)1 |
-0,8* (-1,0, -0,6)b |
-0,3* (-0,6, -0,1)b |
|||
|
N |
153 |
161 |
159 |
166 |
159 |
|
Pacientes (%) que lograron valores de HbA1c <7% con un valor basal de HbA1c ≥7% |
96 (63%) |
112 (70%) |
69 (43%) |
63 (38%) |
92 (58%) |
|
N |
161 |
166 |
168 |
165 |
164 |
|
GPA (mg/dl) [mmol/L] |
|||||
|
Nivel basal (media) |
165,9 [9,2] |
163,7 [9,1] |
170,0 [9,4] |
172,6 [9,6] |
169,0 [9,4] |
|
Cambio respecto del nivel basal1 |
-45,5 [-2,5] |
-47,8 [-2,7] |
-32,9 [-1,8] |
-17,2 [-1,0] |
-32,1 [-1,8] |
|
Comparación vs. empagliflozina (IC 95%)1 |
-12,6** (-19,1,-6,0)b [-0,7 (-1,1, -0,3)] |
-14,8** (-21,4,-8,2)b [-0,8 (-1,2, -0,5)] |
|||
|
Comparación vs. metformina (IC 95%)1 |
-28,2** (-35,0,-21,5)b [-1,6 (-1,9, -1,2)] |
-15,6** (-22,3,-8,9)b [-0,9 (-1,2, -0,5)] |
|||
|
N |
161 |
165 |
168 |
166 |
162 |
|
Peso corporal (kg) |
|||||
|
Nivel basal (media) |
82,3 |
83,0 |
83,9 |
82,9 |
83,8 |
|
Cambio respecto del nivel basal1 |
-3,1 |
-4,1 |
-2,7 |
-0,4 |
-1,2 |
|
Comparación vs. metformina (IC 95%)1 |
-2,7** (-3,6, -1,8)b |
-2,8** (-3,8, -1,9)b |
|||
|
a Administrado en dos dosis fraccionadas en partes iguales por día b Población de análisis completa (caso observado) usando MMRM. El modelo de MMRM incluyó: tratamiento, función renal, región, visita, interacción entre visita y tratamiento y valor basal de HbA1c; la GPA incluyó además el valor basal de GPA; el peso incluyó además el valor basal de peso corporal. 1 Media ajustada para el valor basal 2 Los análisis se realizaron sobre el Grupo Completo de Análisis (GCA) utilizando un enfoque de casos observados (OC) *p≤0,0062 para HbA1c; **Análisis de modo exploratorio: p≤0,0002 para GPA y p<0,0001 para peso corporal. |
|||||
|
Tabla 6. Resultados de un estudio de 24 semanas (OC)2 en el que se comparó la empagliflozina 25 mg en combinación con metformina frente a los componentes individuales de la monoterapia |
|||||
|
Empagliflozina 25 mg +metformina 1000 mga |
Empagliflozina 25 mg+ metformina 2000 mga |
Empagliflozina 25 mg una vez al día |
Metformina 1000 mga |
Metformina 2000 mga |
|
|
N |
165 |
169 |
163 |
167 |
162 |
|
HbA1c (%) |
|||||
|
Nivel basal (media) |
8,8 |
8,7 |
8,9 |
8,7 |
8,6 |
|
Cambio respecto del nivel basal1 |
-1,9 |
-2,1 |
-1,4 |
-1,2 |
-1,8 |
|
Comparación vs. empagliflozina (IC 95%)1 |
-0,6* (-0,8, -0,3)b |
-0,7* (-1,0, -0,5)b |
|||
|
Comparación vs. metformina (IC 95%)1 |
-0,8* (-1,0, -0,5)b |
-0,3* (-0,6, -0,1)b |
|||
|
N |
159 |
163 |
158 |
166 |
159 |
|
Pacientes (%) que lograron valores de HbA1c <7% con un valor basal de HbA1c ≥7% |
91 (57%) |
111 (68%) |
51 (32%) |
63 (38%) |
92 (58%) |
|
N |
163 |
167 |
163 |
165 |
164 |
|
GPA (mg/dl) [mmol/l] |
|||||
|
Nivel basal (media) |
171,2 [9,5] |
167,9 [9,3] |
176,9 [9,8] |
172,6 [9,6] |
169,0 [9,4] |
|
Cambio respecto del nivel basal1 |
-44,0 [-2,4] |
-51,0 [-2,8] |
-28,0 [-1,6] |
-17,2 [-1,0] |
-32,1 [-1,8] |
|
Comparación vs. empagliflozina (IC 95%)1 |
-16,0** (-22,8,-9,2)b [-0,9 (-1,3, -0,5)] |
-23,0** (-29,7,-16,3)b [-1,3 (-1,6, -0,9)] |
|||
|
Comparación vs. metformina (IC 95%)1 |
-26,7** (-33,5,-20,0)b [-1,5 (-1,9, -1,1)] |
-18,8** (-25,5,-12,2)b -1,0 (-1,4, -0,7)] |
|||
|
N |
165 |
167 |
162 |
166 |
162 |
|
Peso corporal (kg) |
|||||
|
Nivel basal (media) |
82,9 |
83,7 |
83,4 |
82,9 |
83,8 |
|
Cambio respecto del nivel basal1 |
-3,6 |
-4,3 |
-2,8 |
-0,4 |
-1,2 |
|
Comparación vs. metformina (IC 95%)1 |
-3,1** (-4,1, -2,2)b |
-3,1** (-4,1, -2,2)b |
|||
|
a Administrado en dos dosis fraccionadas en partes iguales por día b Población de análisis completa (caso observado) usando MMRM. El modelo de MMRM incluyó: tratamiento, función renal, región, visita, interacción entre visita y tratamiento y valor basal de HbA1c; la GPA incluyó además el valor basal de GPA; el peso incluyó además el valor basal de peso corporal. 1 Media ajustada para el valor basal 2 Los análisis se realizaron sobre el Grupo Completo de Análisis (GCA) utilizando un enfoque de casos observados (OC) *p≤0,0056 para HbA1c **Análisis de modo exploratorio: p<0,0001 para GPA y p<0,0001 para peso corporal. |
|||||
Empagliflozina como complemento de un tratamiento combinado de metformina y una sulfonilurea: Se realizó un estudio doble ciego, comparativo con placebo de 24 semanas de duración para evaluar la eficacia y la seguridad de empagliflozina en pacientes que no obtuvieron un beneficio terapéutico suficiente con una combinación de metformina y una sulfonilurea. El tratamiento con empagliflozina condujo a mejoras estadísticamente significativas en la HbA1c y en el peso corporal, y también a reducciones clínicamente significativas en los valores de GPA y presión arterial en comparación con placebo (Tabla 7).
En la extensión doble ciego, comparativa con placebo de este estudio, las reducciones de la HbA1c (cambio respecto del nivel basal de -0,74% para empagliflozina 10 mg, -0,72% para empagliflozina 25 mg y -0,03% para placebo), peso corporal (cambio respecto del nivel basal de - 2,44 kg para empagliflozina 10 mg, -2,28 kg para empagliflozina 25 mg y -0,63 kg para el placebo) y presión arterial (PAS: cambio respecto del nivel basal de -3,8 mmHg para empagliflozina 10 mg, -3,7 mmHg para empagliflozina 25 mg y -1,6 mmHg para el placebo, PAD: cambio respecto del nivel basal de -2,6 mmHg para empagliflozina 10 mg, -2,3 mmHg para empagliflozina 25 mg y -1,4 mmHg para el placebo) se mantuvieron hasta la Semana 76.
|
Tabla 7. Resultados de un estudio comparativo con placebo de 24 semanas (LOCF)3 de empagliflozina como tratamiento complementario de un régimen de metformina y una sulfonilurea (Grupo Completo de Análisis) |
|||
|
Empagliflozina como tratamiento complementario de un régimen de metformina y una sulfonilurea |
Placebo |
Empagliflozina 10 mg |
Empagliflozina 25 mg |
|
N |
225 |
225 |
216 |
|
HbA1c (%) |
|||
|
Nivel basal (media) |
8,15 |
8,07 |
8,10 |
|
Cambio respecto del nivel basal1 |
-0,17 |
-0,82 |
-0,77 |
|
Diferencia respecto del placebo1 (IC 97,5%) |
-0,64* (-0,79, -0,49) |
-0,59* (-0,74, -0,44) |
|
|
N |
216 |
209 |
202 |
|
Pacientes (%) que lograron valores de HbA1c <7% con un valor basal de HbA1c ≥7%2 |
9,3 |
26,3 |
32,2 |
|
N |
224 |
225 |
215 |
|
GPA (mg/dl) [mmol/l]2 |
|||
|
Nivel basal (media) |
151,7 [8,42] |
151,0 [8,38] |
156,5 [8,68] |
|
Cambio respecto del nivel basal1 |
5,5 [0,31] |
-23,3 [-1,29] |
-23,3 [-1,29] |
|
Diferencia respecto del placebo1 (IC 95%) |
-28,8* (-34,2, -23,4) [-1,60* (-1,90, -1,30)] |
-28,8* (-34,3, -23,3) [-1,60* (-1,90, -1,29)] |
|
|
N |
225 |
225 |
216 |
|
Peso corporal (kg) |
|||
|
Nivel basal (media) |
76,23 |
77,08 |
77,50 |
|
Cambio respecto del nivel basal1 |
-0,39 |
-2,16 |
-2,39 |
|
Diferencia respecto del placebo1 (IC 97,5%) |
-1,76* (-2,25, -1,28) |
-1,99* (-2,48, -1,50) |
|
|
N |
225 |
225 |
216 |
|
Pacientes (%) que lograron una pérdida de peso de >5%2 |
5,8 |
27,6 |
23,6 |
|
N |
225 |
225 |
216 |
|
PAS (mmHg)2 |
|||
|
Nivel basal (media) |
128,8 |
128,7 |
129,3 |
|
Cambio respecto del nivel basal1 |
-1,4 |
-4,1 |
-3,5 |
|
Diferencia respecto del placebo1 (IC 95%) |
-2,7 (-4,6, -0,8) |
-2,1 (-4,0, -0,2) |
|
|
1 Media ajustada para el valor basal y estratificación. 2 No evaluado para la determinación de la significancia estadística; no forma parte del procedimiento analítico secuencial aplicado para los criterios de valoración secundarios. 3 Extrapolación de la última observación (previa al rescate glucémico) (LOCF) * Valor p <0,0001. |
|||
Empagliflozina como complemento de un tratamiento combinado de pioglitazona (+/- metformina): La eficacia y seguridad de la empagliflozina en combinación con pioglitazona, con o sin metformina (el 75,5% del total de pacientes estaba recibiendo un régimen de base de metformina) se evaluó en un estudio doble ciego, comparativo con placebo, de 24 semanas de duración. La empagliflozina en combinación con pioglitazona (dosis ≥30 mg) con o sin metformina condujo a reducciones estadísticamente significativas en la HbA1c, la glucosa plasmática en ayunas y el peso corporal, y a reducciones clínicamente significativas en la presión arterial, en comparación con placebo (Tabla 8).
En la extensión doble ciego, comparativa con placebo de este estudio, las reducciones de la HbA1c (cambio respecto del nivel basal de -0,61% para empagliflozina 10 mg, -0,70% para empagliflozina 25 mg y -0,01% para placebo), peso corporal (cambio respecto del nivel basal de - 1,47 kg para empagliflozina 10 mg, -1,21 kg para empagliflozina 25 mg y +0,50 kg para el placebo) y presión arterial (PAS: cambio respecto del nivel basal de -1,7 mmHg para empagliflozina 10 mg, -3,4 mmHg para empagliflozina 25 mg y +0,3 mmHg para el placebo, PAD: cambio respecto del nivel basal de -1,3 mmHg para empagliflozina 10 mg, -2,0 mmHg para empagliflozina 25 mg y +0,2 mmHg para el placebo) se mantuvieron hasta la Semana 76.
|
Tabla 8. Resultados de un estudio comparativo con placebo de 24 semanas (LOCF)3 de empagliflozina como tratamiento complementario de un régimen de pioglitazona con o sin metformina (Grupo Completo de Análisis) |
|||
|
Tratamiento complementario de un régimen de pioglitazona +/- metformina |
Placebo |
Empagliflozina 10 mg |
Empagliflozina 25 mg |
|
N |
165 |
165 |
168 |
|
HbA1c (%) |
|||
|
Nivel basal (media) |
8,16 |
8,07 |
8,06 |
|
Cambio respecto del nivel basal1 |
-0,11 |
-0,59 |
-0,72 |
|
Diferencia respecto del placebo1 (IC 97,5%) |
-0,48* (-0,69, -0,27) |
-0,61* (-0,82, -0,40) |
|
|
N |
155 |
151 |
160 |
|
Pacientes (%) que lograron valores de HbA1c <7% con un valor basal de HbA1c ≥7%3 |
7,7 |
23,8 |
30,0 |
|
N |
165 |
163 |
168 |
|
GPA (mg/dl) [mmol/l] |
|||
|
Nivel basal (media) |
151,93 [8,43] |
152,0 [8,44] |
151,86 [8,43] |
|
Cambio respecto del nivel basal1 |
6,47 [0,37] |
-17,0 [-0,94] |
-21,99 [-1,23] |
|
Diferencia respecto del placebo1 (IC 97,5%) |
-23,5* (-31,8, -15,1) [-1,32 (-1,72, -0,91)] |
-28,5* (-36,7, -20,2) [-1,61 (-2,01, -1,21)] |
|
|
N |
165 |
165 |
168 |
|
Peso corporal (kg) |
|||
|
Nivel basal (media) |
78,1 |
77,97 |
78,93 |
|
Cambio respecto del nivel basal1 |
0,34 |
-1,62 |
-1,47 |
|
Diferencia respecto del placebo1 (IC 97,5%) |
-1,95* (-2,64, -1,27) |
-1,81* (-2,49, -1,13) |
|
|
N |
165 |
165 |
168 |
|
Pacientes (%) que lograron una pérdida de peso de >5%3 |
5,5 |
18,8 |
13,7 |
|
N |
165 |
165 |
168 |
|
PAS (mmHg)2, 3 |
|||
|
Nivel basal (media) |
125,7 |
126,5 |
125,9 |
|
Cambio respecto del nivel basal1 |
0,7 |
-3,1 |
-4,0 |
|
Diferencia respecto del placebo1 (IC 95%) |
-3,9 (-6,2, -1,5) |
-4,7 (-7,1, -2,4) |
|
|
1 Media ajustada para el valor basal y estratificación. 2 No evaluado para la determinación de la significancia estadística; no forma parte del procedimiento analítico secuencial aplicado para los criterios de valoración secundarios. 3 Extrapolación de la última observación (previa al rescate glucémico) (LOCF) * Valor p <0,0001. |
|||
Empagliflozina y linagliptina como complemento del tratamiento con metformina: En un estudio de diseño factorial, pacientes que no lograban un control adecuado con metformina, el tratamiento durante 24 semanas con ambas dosis de empagliflozina, 10 mg y 25 mg, administradas junto con linagliptina 5 mg brindó mejoras estadísticamente significativas en los valores de HbA1c y GPA en comparación con linagliptina 5 mg y también en comparación con la empagliflozina 10 o 25 mg. En comparación con linagliptina 5 mg, ambas dosis de empagliflozina más linagliptina 5 mg brindaron reducciones estadísticamente significativas en el peso corporal y la presión arterial. Una mayor proporción de pacientes con un valor basal de HbA1c ≥7,0% y tratados con empagliflozina más linagliptina logró un valor objetivo de HbA1c de <7% en comparación con linagliptina 5 mg (Tabla 9).
Luego de 24 semanas de tratamiento con empagliflozina + linagliptina, tanto la presión arterial sistólica como la diastólica evidenciaron una disminución, que fue de -5,6/-3,6 mmHg (p <0,001 versus linagliptina 5 mg para PAS y PAD) para empagliflozina 25 mg + linagliptina 5 mg y de -4,1/-2,6 mmHg (p <0,05 versus linagliptina 5 mg para PAS, no especif. para PAD) para empagliflozina 10 mg + linagliptina 5 mg. Las reducciones clínicamente significativas en la presión arterial se mantuvieron por 52 semanas, con valores de -3,8/-1,6 mmHg (p <0,05 versus linagliptina 5 mg para PAS y PAD) para empagliflozina 25 mg + linagliptina 5 mg y de -3,1/-1,6 mmHg (p <0,05 versus linagliptina 5 mg para PAS, no especif. para PAD) para empagliflozina 10 mg + linagliptina 5 mg.
Luego de las 24 semanas, el tratamiento de rescate fue usado en 1 (0,7%) paciente tratado con empagliflozina 25 mg/linagliptina 5 mg y en 3 (2,2%) pacientes tratados con empagliflozina 10 mg/linagliptina 5 mg, en comparación con 4 (3,1%) pacientes tratados con linagliptina 5 mg y 6 (4,3%) pacientes tratados con empagliflozina 25 mg y 1 (0,7%) paciente tratado con empagliflozina 10 mg.
|
Tabla 9. Resultados de un estudio comparativo con placebo de 24 semanas (OC) de empagliflozina y linagliptina como combinación a dosis fija como tratamiento complementario de un régimen con metformina (Grupo Completo de Análisis) |
|||||
|
Empagliflozina /linagliptina (25 mg/5 mg) |
Empagliflozina /linagliptina (10 mg/5 mg) |
Empagliflozina 25 mg |
Empagliflozina 10 mg |
Linagliptina 5 mg |
|
|
N |
134 |
135 |
140 |
137 |
128 |
|
HbA1c (%) - 24 semanas |
|||||
|
Nivel basal (media) |
7,9 |
8,0 |
8,0 |
8,0 |
8,0 |
|
Cambio respecto del valor basal (media ajustada) |
-1,2 |
-1,1 |
-0,6 |
-0,7 |
-0,7 |
|
Comparación vs. linagliptina 5 mg (media ajustada) (IC 5%)2 |
-0,5 (-0,7, -0,3)* |
-0,4 (-0,6, -0,2)* |
|||
|
N |
134 |
135 |
140 |
137 |
128 |
|
HbA1c (%) - 52 semanas1 |
|||||
|
Nivel basal (media) |
7,9 |
8,0 |
8,0 |
8,0 |
8,0 |
|
Cambio respecto del valor basal (media ajustada) |
-1,2 |
-1,0 |
-0,7 |
-0,7 |
-0,5 |
|
Comparación vs. linagliptina 5 mg (media ajustada) (IC 95%)2 |
-0,8 (-1,0, -0,6)* |
-0,60 (-0,8, -0,4)* |
|||
|
N |
134 |
135 |
140 |
137 |
128 |
|
Peso corporal - 24 semanas |
|||||
|
Nivel basal (media) en kg |
85 |
87 |
88 |
86 |
85 |
|
Cambio respecto del nivel basal (media ajustada) |
-3,0 |
-2,6 |
-3,2 |
-2,5 |
-0,7 |
|
Comparación vs. linagliptina 5 mg (media ajustada) (IC 95%)4 |
-2,3 (-3,2, -1,4)* |
-1,9 (-2,8, -1,1)* |
|||
|
N |
123 |
128 |
132 |
125 |
119 |
|
Pacientes (%) que lograron valores de HbA1c <7% con un valor basal de HbA1c ≥7% - 24 semanas |
62 |
58 |
33 |
28 |
36 |
|
Comparación vs. linagliptina 5 mg (cociente de probabilidades [OR]) (IC 95%)3 |
3,5 (1,9, 6,4)* |
2,8 (1,6, 5,0)** |
|||
|
1 No evaluado para la determinación de la significancia estadística debido al procedimiento analítico confirmatorio secuencial aplicado. 2 Población de análisis completa (caso observado) usando MMRM. El modelo de MMRM incluyó: tratamiento, función renal, región, visita, interacción entre visita y tratamiento y valor basal de HbA1c. 3 Población de análisis completa con los pacientes que no completaron considerados como fracasos. La regresión logística incluyó: tratamiento, función renal basal, región geográfica y valor basal de HbA1c. 4 Población de Análisis Total usando la extrapolación de la última observación. El modelo ANCOVA incluyó: tratamiento, función renal, región, valor basal de peso corporal y valor basal de HbA1c. * P <0,0001 ** P <0,001 |
|||||
Empagliflozina en pacientes en los que no se logra un control adecuado con metformina y linagliptina: En los pacientes en los que no se logra un control adecuado con metformina y linagliptina 5 mg, el tratamiento durante 24 semanas tanto con GLYXAMBI 10 mg/5 mg como con GLYXAMBI 25 mg/5 mg brindó mejoras estadísticamente significativas en los parámetros de HbA1c, GPA y peso corporal en comparación con el placebo + linagliptina 5 mg. Una mayor cantidad estadísticamente significativa de pacientes con un valor basal de HbA1c ≥7,0% y tratados con ambas dosis de empagliflozina logró un valor objetivo de HbA1c de <7% en comparación con el placebo + linagliptina 5 mg (Tabla 10). Luego de 24 semanas de tratamiento con empagliflozina, tanto la presión arterial sistólica como la diastólica evidenciaron una disminución, que fue de -2,6/-1,1 mmHg (no especif. versus placebo para PAS y PAD) para empagliflozina 25 mg + linagliptina 5 mg y de -1,3/-0,1 mmHg (no especif. versus placebo para PAS y PAD) para empagliflozina 10 mg + linagliptina 5 mg.
Después de 24 semanas, se observó el uso de tratamiento de rescate en 4 (3,6%) pacientes tratados con empagliflozina 25 mg + linagliptina 5 mg y en 2 (1,8%) pacientes tratados con empagliflozina 10 mg + linagliptina 5 mg, en comparación con 13 (12,0%) pacientes tratados con placebo + linagliptina 5 mg.
|
Tabla 10. Parámetros de eficacia con los que se comparó la empagliflozina frente al placebo como tratamiento complementario en pacientes en los que no se logra un control adecuado con metformina y linagliptina 5 mg |
|||
|
Metformina + Linagliptina 5 mg |
|||
|
Empagliflozina 10 mg1 |
Empagliflozina 25 mg1 |
Placebo2 |
|
|
HbA1c (%) - 24 semanas3 |
|||
|
N |
109 |
110 |
106 |
|
Nivel basal (media) |
7,97 |
7,97 |
7,96 |
|
Cambio respecto del nivel basal (media ajustada) |
-0,65 |
-0,56 |
0,14 |
|
Comparación vs. placebo (media ajustada) (IC del 95%)2 |
-0,79 (-1,02, -0,55) P<0,0001 |
-0,70 (-0,93, -0,46) P<0,0001 |
|
|
GPA (mmol/l) - 24 semanas3 |
|||
|
N |
109 |
109 |
106 |
|
Nivel basal (media) |
9,32 |
9,44 |
9,04 |
|
Cambio respecto del nivel basal (media ajustada) |
-1,46 |
-1,75 |
0,34 |
|
Comparación vs. placebo (media ajustada) (IC del 95%) |
-1,80 (-2,31, -1,28) P<0,0001 |
-2,09 (-2,61, -1,57) P<0,0001 |
|
|
Peso corporal-24 semanas3 |
|||
|
N |
109 |
110 |
106 |
|
Nivel basal (media) en kg |
88,4 |
84,4 |
82,3 |
|
Cambio respecto del nivel basal (media ajustada) |
-3,1 |
-2,5 |
-0,3 |
|
Comparación vs. placebo (media ajustada) (IC del 95%)1 |
-2,8 (-3,5, -2,1) P<0,0001 |
-2,2 (-2,9, -1,5) P<0,0001 |
|
|
Pacientes (%) que logran un valor de HbA1c <7% con un valor basal de HbA1c ≥7% -24 semanas4 |
100 |
107 |
100 |
|
N |
|||
|
Pacientes (%) que logran un valor de A1c <7% |
37,0 |
32,7 |
17,0 |
|
Comparación vs. placebo (Cociente de probabilidades) (IC del 95%)5 |
4,0 (1,9, 8,7) P=0,0004 |
2,9 (1,4, 6,1) p=0,0061 |
|
|
1Los pacientes aleatorizados a los grupos de empagliflozina 10 mg o 25 mg estaban recibiendo GLYXAMBI® 10 mg/5 mg o 25 mg/5 mg con tratamiento de base de metformina. 2Los pacientes aleatorizados al grupo de placebo recibían el placebo más linagliptina 5 mg con tratamiento de base de metformina. 3El modelo MMRM incluye valor basal de HbA1c, valor basal de TFGe (MDRD), región geográfica, visita, e interacción entre visita y tratamiento, sobre la base del GCA (OC). Para los valores de GPA, también se incluye el valor basal de GPA. Para el valor del peso, también se incluye el valor basal del peso. 4No evaluado para la determinación de la significancia estadística; no forma parte del procedimiento analítico secuencial para los criterios de valoración secundarios. 5La regresión logística incluye valor basal de HbA1c, valor basal de TFGe (MDRD), región geográfica y tratamiento, sobre la base del GCA (NCF); basado en pacientes con valores de HbA1c de 7% o más en el nivel basal. |
|||
En un subgrupo preespecificado de pacientes con un nivel basal de HbA1c de 8,5% o más, la reducción respecto del nivel basal en el parámetro HbA1c lograda con empagliflozina 25 mg + linagliptina 5 mg fue del -1,3% a las 24 semanas (p<0,0001 versus placebo + linagliptina 5 mg) y con empagliflozina 10 mg + linagliptina 5 mg del -1,3% a las 24 semanas (p<0,0001 versus placebo+ linagliptina 5 mg).
Datos de 2 años de empagliflozina como tratamiento complementario de la metformina en comparación con la glimepirida: En un estudio en el que se comparó la eficacia y la seguridad de empagliflozina 25 mg versus la glimepirida (4 mg) en pacientes con un control glucémico inadecuado con monoterapia de metformina, el tratamiento con un régimen diario de empagliflozina condujo a una reducción superior en los valores de HbA1c, y a una reducción clínicamente significativa en los valores de GPA, en comparación con la glimepirida (Tabla 11). La empagliflozina en un régimen diario condujo a una reducción estadísticamente significativa en el peso corporal, la presión arterial sistólica y diastólica (cambio respecto del nivel basal en la PAD de -1,8 mmHg para empagliflozina y de +0,9 mmHg para glimepirida, p <0,0001).
El tratamiento con empagliflozina condujo a una proporción significativamente menor, desde el punto de vista estadístico, de pacientes con eventos hipoglucémicos en comparación con la glimepirida (2,5% para empagliflozina, 24,2% para glimepirida, p<0,0001).
|
Tabla 11. Resultados de un estudio con control activo de 104 semanas (LOCF)4 en el que se comparó la empagliflozina frente a la glimepirida como tratamiento complementario a un régimen de metformina (Grupo Completo de Análisis) |
||
|
Empagliflozina como tratamiento complementario de un régimen de metformina en comparación con la glimepirida |
Empagliflozina 25 mg |
Glimepirida (hasta 4 mg) |
|
N |
765 |
780 |
|
HbA1c (%) |
||
|
Nivel basal (media) |
7,92 |
7,92 |
|
Cambio respecto del nivel basal1 |
-0,66 |
-0,55 |
|
Diferencia respecto de la glimepirida1 (IC 97,5%) |
-0,11* (-0,20, -0,01) |
|
|
N |
690 |
715 |
|
Pacientes (%) que lograron valores de HbA1c <7% con un valor basal de HbA1c ≥7%2 |
33,6 |
30,9 |
|
N |
||
|
GPA (mg/dl) [mmol/l] |
||
|
Nivel basal (media) |
150,00 |
149,82 |
|
Cambio respecto del nivel basal1 |
-15,36 |
-2,98 |
|
Diferencia respecto de la glimepirida1 (IC 95%) |
-12,37** (-15,47, -9,27) |
|
|
N |
765 |
780 |
|
Peso corporal (kg) |
||
|
Nivel basal (media) |
82,52 |
83,03 |
|
Cambio respecto del nivel basal1 |
-3,12 |
1,34 |
|
Diferencia respecto de la glimepirida1 (IC 97,5%) |
-4,46** (-4,87, -4,05) |
|
|
N |
765 |
780 |
|
Pacientes (%) que lograron una pérdida de peso de >5%2 |
27,5 |
3,8% |
|
N |
765 |
780 |
|
PAS (mmHg)3 |
||
|
Nivel basal (media) |
133,4 |
133,5 |
|
Cambio respecto del nivel basal1 |
-3,1 |
2,5 |
|
Diferencia respecto de la glimepirida1 (IC 97,5%) |
-5,6** (-7,0, -4,2) |
|
|
1Media ajustada para el valor basal y estratificación. 2 No evaluado para la determinación de la significancia estadística; no forma parte del procedimiento analítico secuencial aplicado para los criterios de valoración secundarios. 3 LOCF, los valores posteriores a un rescate antihipertensivo se censuraron 4 Extrapolación de la última observación (previa al rescate glucémico) (LOCF) * Valor p < 0,0001 para no inferioridad, y valor p = 0,0153 para superioridad. ** Valor p <0,0001. |
||
Empagliflozina como tratamiento complementario de un régimen de insulina basal: La eficacia y seguridad de la empagliflozina como tratamiento complementario de un régimen de insulina basal con o sin administración de un tratamiento concomitante de metformina y/o una sulfonilurea (el 79,8% del total de pacientes estaba recibiendo tratamiento de base con metformina) se evaluó en un estudio doble ciego, comparativo con placebo, de 78 semanas de duración. Durante las 18 semanas iniciales, la dosis de insulina se debía mantener estable, pero se ajustó para lograr un valor de GPA <110 mg/dl en las 60 semanas siguientes.
En la semana 18, la empagliflozina brindó una mejora estadísticamente significativa en los valores de HbA1c en comparación con placebo. Una mayor proporción de pacientes con un valor basal de HbA1c ≥7,0% logró un valor objetivo de HbA1c de <7% en comparación con placebo. A las 78 semanas, la empagliflozina brindaba una reducción estadísticamente significativa de los valores de HbA1c y una reducción de la necesidad de insulina en comparación con placebo (Tabla 12).
En la semana 78, la empagliflozina brindó una reducción en los valores de GPA de -10,51 mg/dl [- 0,58 mmol/l] para empagliflozina 10 mg, de -17,43 mg/dl [0,3 mmol/l] para empagliflozina 25 mg y de -5,48 mg/dl [0,97 mmol/l] para el placebo), en el peso corporal (-2,47 kg para empagliflozina 10 mg, -1,96 kg para empagliflozina 25 mg y +1,16 kg para placebo, p <0,0001), en la presión arterial (PAS: -4,1 mmHg para empagliflozina 10 mg, -2,4 mmHg para empagliflozina 25 mg y +0,1 mmHg para placebo, PAD: -2,9 mmHg para empagliflozina 10 mg, -1,5 mmHg para empagliflozina 25 mg y -0,3 mmHg para placebo).
|
Tabla 12. Resultados de un estudio comparativo con placebo de 18 y 78 semanas (LOCF)2 de empagliflozina como tratamiento complementario de un régimen de insulina basal con o sin metformina y/o una sulfonilurea (Grupo Completo de Análisis; Pacientes que Completaron) |
|||
|
Tratamiento complementario de un régimen de insulina basal +/- metformina o una sulfonilurea |
Placebo |
Empagliflozina 10 mg |
Empagliflozina 25 mg |
|
N |
125 |
132 |
117 |
|
HbA1c (%) en la Semana 18 |
|||
|
Nivel basal (media) |
8,10 |
8,26 |
8,34 |
|
Cambio respecto del nivel basal1 |
-0,01 |
-0,57 |
-0,71 |
|
Diferencia respecto del placebo1 (IC 97,5%) |
-0,56* (-0,78, -0,33) |
-0,70* (-0,93, -0,47) |
|
|
N |
112 |
127 |
110 |
|
HbA1c (%) en la Semana 78 |
|||
|
Nivel basal (media) |
8,09 |
8,27 |
8,29 |
|
Cambio respecto del nivel basal1 |
-0,02 |
-0,48 |
-0,64 |
|
Diferencia respecto del placebo1 (IC 97,5%) |
-0,46* (-0,73, -0,19) |
-0,62* (-0,90, -0,34) |
|
|
N |
112 |
127 |
110 |
|
Dosis de insulina basal (UI/día) en la semana 78 |
|||
|
Nivel basal (media) |
47,84 |
45,13 |
48,43 |
|
Cambio respecto del nivel basal1 |
5,45 |
-1,21 |
-0,47 |
|
Diferencia respecto del placebo1 (IC 97,5%) |
-6,66*** (-11,56, -1,77) |
-5,92*** (-11,00, -0,85) |
|
|
1 Media ajustada para el valor basal y estratificación. 2 Extrapolación de la última observación (previa al rescate glucémico) (LOCF) * Valor p <0,0001. *** Valor p <0,01. |
|||
Empagliflozina como tratamiento complementario de un régimen de insulina MDI y metformina: La eficacia y seguridad de la empagliflozina como tratamiento complementario de un régimen de dosis diarias múltiples de insulina con o sin administración de un tratamiento concomitante de metformina (el 71,0% del total de pacientes estaba recibiendo tratamiento de base con metformina) se evaluó en un estudio doble ciego, comparativo con placebo, de 52 semanas de duración. Durante las primeras 18 semanas y las últimas 12 semanas, la dosis de insulina se mantuvo estable, pero se ajustó para lograr niveles de glucosa preprandial <100 mg/dl [5,5 mmol/l] y niveles de glucosa posprandial <140 mg/dl [7,8 mmol/l] entre las semanas 19 y 40.
En la semana 18, la empagliflozina brindó una mejora estadísticamente significativa en los valores de HbA1c en comparación con placebo (Tabla 13). Una mayor proporción de pacientes con un valor basal de HbA1c ≥7,0% (19,5% para empagliflozina 10 mg, 31,0% para empagliflozina 25 mg) logró un valor objetivo de HbA1c <7% en comparación con placebo (15,1%).
En la semana 52, el tratamiento con empagliflozina condujo a un descenso estadísticamente significativo en los valores de HbA1c, a una reducción de la necesidad de insulina en comparación con placebo así como una reducción en los valores de GPA (cambio respecto del nivel basal de -0,3 mg/dl [-0,02 mmol/l] para placebo, -19,7 mg/dl [-1,09 mmol/l] para empagliflozina 10 mg, y -23,7 mg/dl [-1,31 mmol/l] para empagliflozina 25 mg), peso corporal y presión arterial (PAS: cambio respecto del nivel basal de -2,6 mmHg para placebo, -3,9 mmHg para empagliflozina 10 mg y 4,0 mmHg para empagliflozina 25 mg, PAD: cambio respecto del nivel basal de -1,0 mmHg para el placebo, -1,4 mmHg para empagliflozina 10 mg y -2,6 mmHg para empagliflozina 25 mg).
|
Tabla 13. Resultados obtenidos a las 18 y 52 (LOCF)5 semanas en un estudio comparativo con placebo de empagliflozina como tratamiento complementario de dosis diarias múltiples de insulina con metformina2 |
|||
|
Empagliflozina como complemento del tratamiento con insulina + metformina |
Placebo |
Empagliflozina 10 mg |
Empagliflozina 25 mg |
|
N |
188 |
186 |
189 |
|
HbA1c (%) en la Semana 18 |
|||
|
Nivel basal (media) |
8,33 |
8,39 |
8,29 |
|
Cambio respecto del nivel basal1 |
-0,50 |
-0,94 |
-1,02 |
|
Diferencia respecto del placebo1 (IC 97,5%) |
-0,44* (-0,61, -0,27) |
-0,52* (-0,69, -0,35) |
|
|
N |
115 |
119 |
118 |
|
HbA1c (%) en la semana 523 |
|||
|
Nivel basal (media) |
8,25 |
8,40 |
8,37 |
|
Cambio respecto del nivel basal1 |
-0,81 |
-1,18 |
-1,27 |
|
Diferencia respecto del placebo1 (IC 97,5%) |
-0,38** (-0,62, -0,13) |
-0,46* (-0,70, -0,22) |
|
|
N |
113 |
118 |
118 |
|
Pacientes (%) que lograron valores de <7% con un valor basal de HbA1c ≥7% en la semana 524 |
26,5 |
39,8 |
45,8 |
|
N |
188 |
186 |
189 |
|
GPA (mg/dl) [mmol/l] en la semana 525 |
|||
|
Nivel basal (media) |
151,6 [8,41] |
159,1 [8,83] |
150,3 [8,34] |
|
Cambio respecto del nivel basal1 |
-0,3 [-0,02] |
-19,7 [-1,09] |
-23,7 [-1,31] |
|
Diferencia respecto del placebo1 (IC 95%) |
-19,3 (-27,9, -10,8) [-1,07(-1,55, -0,6)] |
-23,4 (-31,8, -14,9) [-1,30(-1,77, -0,83)] |
|
|
N |
115 |
118 |
117 |
|
Dosis de insulina (UI/día) en la semana 523 |
|||
|
Nivel basal (media) |
89,94 |
88,57 |
90,38 |
|
Cambio respecto del nivel basal1 |
10,16 |
1,33 |
-1,06 |
|
Diferencia respecto del placebo1 (IC 97,5%) |
-8,83** (-15,69, - 1,97) |
-11,22**(-18,09, -4,36) |
|
|
N |
115 |
119 |
118 |
|
Peso corporal (kg) en la semana 523 |
|||
|
Nivel basal (media) |
96,34 |
96,47 |
95,37 |
|
Cambio respecto del nivel basal1 |
0,44 |
-1,95 |
-2,04 |
|
Diferencia respecto del placebo1 (IC 97,5%) |
-2,39* (-3,54, -1,24) |
-2,48* (-3,63, -1,33) |
|
|
N |
188 |
186 |
189 |
|
PAS (mmHg)5 |
|||
|
Nivel basal (media) |
132,6 |
134,2 |
132,9 |
|
Cambio respecto del nivel basal1 |
-2,6 |
-3,9 |
-4,0 |
|
Diferencia respecto del placebo1,4 (IC 95%) |
-1,4 (-3,6, 0,9) |
-1,4 (-3,7, 0,8) |
|
|
1 Media ajustada para el valor basal y estratificación. 2 Semana 18: GCA; semana 52: PPS (Conjunto por Protocolo) - Pacientes que Completaron - 52 3 Semana 19-40: régimen de tratamiento orientado al objetivo para el ajuste de la dosis de insulina para lograr los niveles objetivo de glucosa predefinidos (preprandial <100 mg/dl (5,5 mmol/l), posprandial <140 mg/dl (7,8 mmol/l) 4No evaluado para la determinación de la significancia estadística; no forma parte del procedimiento analítico secuencial aplicado para los criterios de valoración secundarios. 5 Extrapolación de la última observación (previa al rescate glucémico) (LOCF) 6 Semana 52: GCA * Valor p <0,0001. ** Valor p <0,001. |
|||
Régimen de dos dosis diarias de empagliflozina versus un régimen de una dosis diaria de empagliflozina como complemento del tratamiento con metformina: La eficacia y seguridad de empagliflozina dos veces al día versus una vez al día (dosis diaria de 10 mg y 25 mg) como tratamiento complementario en pacientes con un control glucémico insuficiente con la monoterapia de metformina fue evaluada en un estudio doble ciego, comparativo con placebo de 16 semanas de duración. Todos los tratamientos con empagliflozina condujeron a reducciones significativas en los valores de HbA1c respecto del nivel basal (media total 7,8%) al cabo de 16 semanas de tratamiento en comparación con placebo. Los regímenes de dos dosis diarias de empagliflozina condujeron a reducciones comparables en los valores de HbA1c en comparación con los regímenes de una dosis diaria con una diferencia entre los tratamientos en términos de reducciones en los valores de HbA1c entre el nivel basal y la semana 16 de -0,02% (IC 95% -0,16, 0,13) para empagliflozina 5 mg dos veces al día vs. 10 mg una vez al día, y de -0,11% (IC 95% -0,26, 0,03) para empagliflozina 12,5 mg dos veces al día vs. 25 mg una vez al día.
Glucosa posprandial a las 2 horas: El tratamiento con empagliflozina como tratamiento complementario a un régimen de metformina o de metformina más una sulfonilurea condujo a una mejora clínicamente significativa en los valores de glucosa posprandial a las 2 horas (prueba de tolerancia a los alimentos) a las 24 semanas (tratamiento complementario de un régimen de metformina, placebo (n=57): +5,9 mg/dl, empagliflozina 10 mg (n=52): -46,0 mg/dl, empagliflozina 25 mg (n=58): -44,6 mg/dl; tratamiento complementario de un régimen de metformina más una sulfonilurea, placebo (n=35): -2,3 mg/dl, empagliflozina 10 mg (n=44): -35,7 mg/dl, empagliflozina 25 mg (n=46): -36,6 mg/dl).
Pacientes con valores basales de HbA1c ≥9%
En un análisis preespecificado de sujetos con valores basales de HbA1c ≥9,0%, el tratamiento con empagliflozina 10 mg o 25 mg como tratamiento complementario de un régimen de metformina condujo a reducciones estadísticamente significativas en los valores de HbA1c en la Semana 24 (media ajustada para el cambio respecto del nivel basal de -1,49% para empagliflozina 25 mg, de - 1,40% para empagliflozina 10 mg y de -0,44% para placebo).
Peso corporal: En un análisis preespecificado de datos combinados de 4 estudios comparativos con placebo, el tratamiento con empagliflozina (el 68% del total de pacientes estaba recibiendo un régimen de base de metformina) condujo a un descenso del peso corporal en comparación con el placebo en la semana 24 (-2,04 kg para empagliflozina 10 mg, -2,26 kg para empagliflozina 25 mg y -0,24 kg para placebo) que se mantuvo hasta la semana 52 (-1,96 kg para empagliflozina 10 mg, -2,25 kg para empagliflozina 25 mg y -0,16 kg para placebo).
Presión arterial: La eficacia y seguridad de la empagliflozina se evaluó en un estudio doble ciego, comparativo con placebo de 12 semanas de duración en pacientes con diabetes tipo 2 e hipertensión arterial que estaban recibiendo diferentes tratamientos antidiabéticos (el 67,8% estaba siendo tratado con metformina con o sin otro fármaco antidiabético, incluyendo insulina) y hasta 2 tratamientos antihipertensivos (Tabla 14). El tratamiento con empagliflozina en un régimen de una toma diaria condujo a una mejora estadísticamente significativa en los valores de HbA1c y en los valores medios de 24 horas de presión arterial sistólica y diastólica determinados por monitoreo ambulatorio de la presión arterial. El tratamiento con empagliflozina se tradujo en reducciones en los valores de PAS en el paciente sentado (cambio respecto del nivel basal de -0,67 mmHg para placebo, de -4,60 mmHg para empagliflozina 10 mg y de -5,47 mmHg para la empagliflozina 25 mg) y en los valores de PAD en el paciente sentado (cambio respecto del nivel basal de -1,13 mmHg para placebo, de -3,06 mmHg para empagliflozina 10 mg y de -3,02 mmHg para la empagliflozina 25 mg).
|
Tabla 14. Resultados de un estudio comparativo con placebo de 12 semanas (LOCF)3 de empagliflozina en pacientes con diabetes tipo 2 con presión arterial no controlada (Grupo Completo de Análisis) |
|||
|
Placebo |
Empagliflozina 10 mg |
Empagliflozina 25 mg |
|
|
N |
271 |
276 |
276 |
|
HbA1c (%) en la semana 12 |
|||
|
Nivel basal (media) |
7,90 |
7,87 |
7,92 |
|
Cambio respecto del nivel basal1 |
0,03 |
-0,59 |
-0,62 |
|
Diferencia respecto del placebo1 (IC 95%) |
-0,62* (-0,72, -0,52) |
-0,65* (-0,75, -0,55) |
|
|
PAS 24 horas en la semana 122 |
|||
|
Nivel basal (media) |
131,72 |
131,34 |
131,18 |
|
Cambio respecto del nivel basal1 |
0,48 |
-2,95 |
-3,68 |
|
Diferencia respecto del placebo1 (IC 95%) |
-3,44* (-4,78, -2,09) |
-4,16* (-5,50, -2,83) |
|
|
PAD 24 horas en la semana 122 |
|||
|
Nivel basal (media) |
75,16 |
75,13 |
74,64 |
|
Cambio respecto del nivel basal1 |
0,32 |
-1,04 |
-1,40 |
|
Diferencia respecto del placebo1 (IC 95%) |
-1,36** (-2,15, -0,56) |
-1,72* (-2,51, -0,93) |
|
|
1 Media ajustada para el valor basal y estratificación. 2 Extrapolación de la última observación (previa al rescate antihipertensivo) (LOCF) LOCF, los valores posteriores al rescate antihipertensivo se censuraron. 3 Extrapolación de la última observación (previa al rescate glucémico) (LOCF) * Valor p <0,0001. ** Valor p =0,0008. |
|||
En un análisis preespecificado de datos combinados de 4 estudios comparativos con placebo, el tratamiento con empagliflozina (el 68% del total de pacientes estaba recibiendo un régimen de base de metformina) condujo a un descenso en la presión arterial sistólica (empagliflozina 10 mg -3,9 mmHg, empagliflozina 25 mg -4,3 mmHg) en comparación con placebo (-0,5 mmHg) y a un descenso en la presión arterial diastólica (empagliflozina 10 mg -1,8 mmHg, empagliflozina 25 mg -2,0 mmHg) en comparación con placebo (-0,5 mmHg), en la semana 24, que se mantuvo hasta la semana 76.
Parámetros de laboratorio:
Incremento del hematocrito: En un análisis de seguridad de todos los ensayos con metformina como tratamiento de base, los cambios promedio desde la línea de base en el hematocrito fueron de 3,6% y 4,0% para empagliflozina 10 mg y 25 mg, respectivamente, en comparación con el 0% para el placebo. En el estudio EMPA-REG OUTCOME®, los valores de hematocrito volvieron hacia los valores basales después de un período de seguimiento de 30 días después de suspender el tratamiento.
Aumento de lípidos en suero: En un análisis de seguridad de todos los ensayos con metformina como tratamiento de base, los aumentos promedio respecto al inicio para empagliflozina 10 mg y 25 mg versus placebo, respectivamente, fueron: colesterol total un 5,0% y 5,2% versus 3,7%; HDL-colesterol 4,6% y 2,7%
frente a -0,5%; LDL-colesterol en un 9,1% y un 8,7% frente a 7,8%; Los triglicéridos 5,4% y 10,8% frente a 12,1%.
Resultado cardiovascular: El estudio EMPA-REG OUTCOME® es un ensayo multinacional, multicéntrico, aleatorizado, doble ciego, comparativo con placebo para investigar el efecto de la empagliflozina como complemento del tratamiento estándar en la reducción de eventos cardiovasculares en pacientes con diabetes mellitus tipo 2 y uno o más factores de riesgo, incluidas la enfermedad coronaria, la enfermedad arterial periférica, los antecedentes de infarto de miocardio (IM) y los antecedentes de accidente cerebrovascular. El criterio de valoración primario fue el tiempo transcurrido hasta el primer evento en el compuesto de muerte CV, IM no fatal o accidente cerebrovascular no fatal (Episodios Cardiovasculares Graves [MACE-3]). Se incluyeron criterios de valoración predeterminados adicionales para abordar los resultados clínicamente relevantes evaluados en forma exploratoria, entre los cuales se encuentran: muerte CV, el compuesto de insuficiencia cardíaca que requiere hospitalización o muerte CV, mortalidad por todas las causas y el compuesto de nuevo caso o empeoramiento de nefropatía.
Se administró un tratamiento con empagliflozina a 7020 pacientes (empagliflozina 10 mg: 2345, empagliflozina 25 mg: 2342, placebo: 2333) y se realizó un seguimiento por una mediana de 3,1 años. La población estuvo compuesta en un 72,4% por pacientes caucásicos, un 21,6% por asiáticos y un 5,1% por negros. La edad promedio era de 63 años y el 71,5% eran hombres. Al inicio, aproximadamente el 81% de los pacientes recibían tratamiento con inhibidores del sistema renina-angiotensina, el 65% con beta-bloqueantes, el 43% con diuréticos, el 89% con anticoagulantes y el 81% con medicación hipolipemiante. Aproximadamente el 74% de los pacientes recibían tratamiento con metformina al inicio, el 48% con insulina y el 43% con sulfonilurea.
Aproximadamente la mitad de los pacientes (52,2%) tenían niveles de TFGe de 60-90 ml/min/1,73 m2, el 17,8% de 45-60 ml/min/1,73 m2 y el 7,7% de 30-45 ml/min/1,73 m2. La media de PAS fue de 136 mmHg, PAD 76 mmHg, LDL 86 mg/dl, HDL 44 mg/dl y el cociente albúmina/creatinina en orina (UACR) 175 mg/g al inicio.
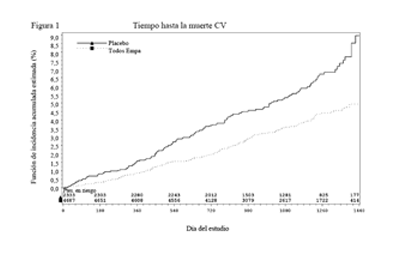
Reducciones en el riesgo de muerte CV y mortalidad global: La empagliflozina es superior a placebo en la reducción del criterio de valoración primario compuesto de muerte cardiovascular, IM no fatal o accidente cerebrovascular no fatal. El efecto del tratamiento reflejó una reducción en la muerte cardiovascular sin cambios significativos en el IM no fatal l o el accidente cerebrovascular no fatal (Tabla 15 y Figura 1).
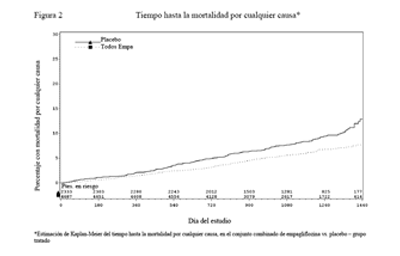
La empagliflozina también mejoró la sobrevida global (Tabla 15 y Figura 2), como resultado de una reducción en la muerte cardiovascular con la administración de empagliflozina. No se observaron diferencias estadísticamente significativas entre la empagliflozina y placebo en la mortalidad no cardiovascular.
|
Tabla 15. Efecto del tratamiento para el criterio de valoración compuesto primario, sus componentes y la mortalidad (Grupo Tratado*) |
||
|
Placebo |
Empagliflozina (10 y 25 mg, combinados) |
|
|
N |
2333 |
4687 |
|
Tiempo hasta la primera aparición de muerte CV, IM no fatal o accidente cerebrovascular no fatal N (%) |
282 (12,1) |
490 (10,5) |
|
Razón de riesgos instantáneos vs. placebo (IC 95,02%)** |
0,86 (0,74, 0,99) |
|
|
Valor p para superioridad |
0,0382 |
|
|
Muerte cardiovascular N (%) |
137 (5,9) |
172 (3,7) |
|
Razón de riesgos instantáneos vs. placebo (IC 95%) |
0,62 (0,49; 0,77) |
|
|
Valor p |
<0,0001 |
|
|
IM no fatal N (%) |
121 (5,2) |
213 (4,5) |
|
Razón de riesgos instantáneos vs. placebo (IC 95%) |
0,87 (0,70; 1,09) |
|
|
Valor p |
0,2189 |
|
|
Accidente cerebrovascular no fatal N (%) |
60 (2,6) |
150 (3,2) |
|
Razón de riesgos instantáneos vs. placebo (IC 95%) |
1,24 (0,92; 1,67) |
|
|
Valor p |
0,1638 |
|
|
Mortalidad por cualquier causa N (%) |
194 (8,3) |
269 (5,7) |
|
Razón de riesgos instantáneos vs. placebo (IC 95%) |
0,68 (0,57; 0,82) |
|
|
Valor p |
<0,0001 |
|
|
Mortalidad no CV N (%) |
57 (2,4) |
97 (2,1) |
|
Razón de riesgos instantáneos vs. placebo (IC 95%) |
0,84 (0,60; 1,16) |
|
|
* es decir, pacientes que habían recibido por lo menos una dosis del medicamento en estudio ** dado que los datos del estudio se incluyeron en un análisis intermedio, se aplicó un intervalo de confianza bilateral del 95,02% que correspondió a un valor p menor a 0,0498 para significancia. |
||
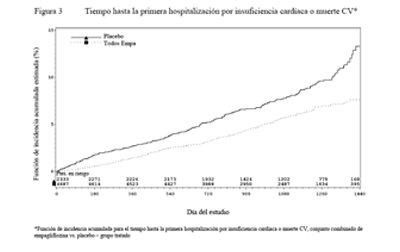
Reducciones en el riesgo de insuficiencia cardíaca que requiere hospitalización o muerte CV: La empagliflozina es superior a placebo en la reducción del riesgo de hospitalización por insuficiencia cardíaca y muerte cardiovascular u hospitalización por insuficiencia cardíaca (Tabla 16 y Figura 3).
|
Tabla 16. Efecto del tratamiento en la hospitalización por insuficiencia cardíaca o muerte cardiovascular (excluido el ACV fatal) (Grupo Tratado*) |
||
|
Placebo |
Empagliflozina** (10 y 25 mg, combinados) |
|
|
N |
2333 |
4687 |
|
Insuficiencia cardíaca que requiere hospitalización o muerte cardiovascular (excluido el ACV fatal) N (%)*** |
198 (8,5) |
265 (5,7) |
|
HR (IC 95%) |
0,66 (0,55; 0,79) |
|
|
Valor p |
<0,0001 |
|
|
Insuficiencia cardíaca que requiere hospitalización N (%) |
95 (4,1) |
126 (2,7) |
|
HR (IC 95%) |
0,65 (0,50; 0,85) |
|
|
Valor p |
0,0017 |
|
|
Muerte cardiovascular (excluido el ACV fatal) N (%) |
126 (5,4) |
156 (3,3) |
|
HR (IC 95%) |
0,61 (0,48; 0,77) |
|
|
Valor p |
<0,0001 |
|
|
* es decir, pacientes que habían recibido por lo menos una dosis del medicamento en estudio ** la empagliflozina 10 mg y 25 mg demostró resultados consistentes *** tiempo hasta el primer evento |
||
Los beneficios cardiovasculares observados de la empagliflozina fueron consistentes en todos los subgrupos que se presentan en la Figura 4.
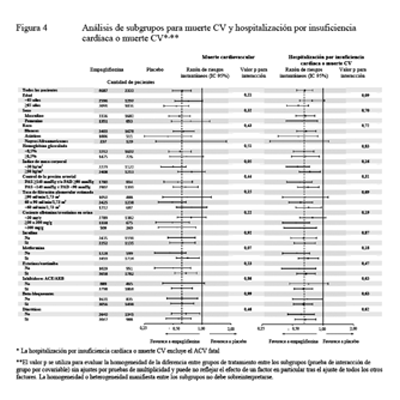
En el subgrupo de pacientes que recibían metformina al inicio, los efectos en los resultados CV fueron consistentes con los resultados observados en toda la población del estudio EMPA REG OUTCOME®.
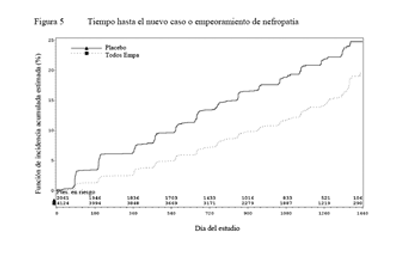
Enfermedad renal diabética: En la población del estudio EMPA-REG OUTCOME®, el riesgo de nuevos casos o empeoramiento de nefropatía (definidos como el comienzo de macroalbuminuria, duplicación de la creatinina sérica e inicio de tratamiento de reemplazo renal [es decir, hemodiálisis]) se redujo significativamente en el grupo de empagliflozina en comparación con placebo (Tabla 17 y Figura 5).
Comparada con placebo, la empagliflozina demostró una mayor presentación de normo o microalbuminuria sostenida en pacientes con macroalbuminuria al inicio (HR 1,82, IC 95%: 1,40; 2,37).
|
Tabla 17. Tiempo hasta el nuevo caso o empeoramiento de nefropatía (Grupo Tratado*) |
||
|
Placebo |
Empagliflozina (10 y 25 mg, combinados) |
|
|
N |
2061 |
4124 |
|
Nuevo caso o empeoramiento de nefropatía N (%) |
388 (18,8) |
525 (12,7) |
|
HR (IC 95%) |
0,61 (0,53, 0,70) |
|
|
Valor p |
<0,0001 |
|
|
N |
2323 |
4645 |
|
Duplicación del nivel de creatinina sérica** N (%) |
60 (2,6) |
70 (1,5) |
|
HR (IC 95%) |
0,56 (0,39, 0,79) |
|
|
Valor p |
0,0009 |
|
|
N |
2033 |
4091 |
|
Nueva presentación de macroalbuminuria*** N (%) |
330 (16,2) |
459 (11,2) |
|
HR (IC 95%) |
0,62 (0,54, 0,72) |
|
|
Valor p |
<0,0001 |
|
|
N |
2333 |
4687 |
|
Inicio o continuación de tratamiento de reemplazo renal N (%) |
14 (0,6) |
13 (0,3) |
|
HR (IC 95%) |
0,45 (0,21, 0,97) |
|
|
Valor p |
0,0409 |
|
|
N |
2333 |
4687 |
|
Muerte por enfermedad renal N (%)**** |
0 |
3 (0,1) |
|
* Es decir, pacientes que habían recibido por lo menos una dosis del medicamento en estudio ** Acompañada por una TFGe ≤45 ml/min/1,73 m2 *** Cociente albúmina/creatinina en orina >300 mg/g **** Dado el bajo índice de eventos, el HR no se calculó |
||
El tratamiento con empagliflozina preservó la TFGe y el aumento de la TFGe durante las 4 semanas de seguimiento posteriores al tratamiento. No obstante, el grupo de placebo demostró una reducción gradual en la TFGe durante el curso del estudio sin cambios adicionales durante el seguimiento de 4 semanas (véase la Figura 6).

En el subgrupo de pacientes que recibían metformina al inicio, los efectos en estos resultados renales fueron consistentes con los resultados observados en toda la población del estudio EMPA REG OUTCOME®.
Estudio de QTc exhaustivo: En un estudio aleatorizado, comparativo con placebo, con comparador activo, de diseño cruzad en el que se evaluaron 30 sujetos sanos, no se observó prolongación del QTc con la dosis de 25 mg ni con la dosis de 200 mg de empagliflozina.
FARMACOCINÉTICA:
JARDIANCE DUO®
Los resultados de los estudios de bioequivalencia realizados en sujetos sanos demostraron que los comprimidos combinados de JARDIANCE DUO® (empagliflozina/clorhidrato de metformina) de 5 mg/500 mg, 5 mg/850 mg, 5 mg/1000 mg, 12,5 mg/500 mg, 12,5 mg/850 mg y 12,5 mg/1000 mg son bioequivalentes a la coadministración de las correspondientes dosis de empagliflozina y metformina como comprimidos individuales.
La administración de 12,5 mg empagliflozina/1000 mg metformina en estado posprandial condujo a una reducción del 9% en el AUC (área bajo la curva) y a una reducción del 28% en la Cmax correspondientes a empagliflozina, en comparación con la administración en ayunas. En el caso de la metformina, el AUC se redujo un 12% y la Cmax se redujo un 26% en comparación con la administración en ayunas. El efecto observado de los alimentos en relación con la empagliflozina y con la metformina no se considera relevante desde el punto de vista clínico. No obstante, dado que se recomienda que la metformina se administre junto con las comidas, también se propone la administración junto con las comidas en el caso de JARDIANCE DUO®.
Los datos que se brindan a continuación son hallazgos de estudios realizados con empagliflozina o metformina en forma individual.
Empagliflozina:
Absorción: La farmacocinética de empagliflozina ha sido ampliamente caracterizada en voluntarios sanos y en pacientes con DMT2. Tras la administración por vía oral, la empagliflozina se absorbió rápidamente, con concentraciones plasmáticas pico alcanzadas en una mediana de tmax de 1,5 horas posdosis. A partir de ese momento, las concentraciones plasmáticas se redujeron siguiendo un patrón bifásico con una fase de distribución rápida y una fase terminal relativamente lenta. Los valores plasmáticos medios en estado de equilibrio dinámico para AUC y Cmax fueron 1870 nmol.h y 259 nmol/l con empagliflozina 10 mg y 4740 nmol.h/l y 687 nmol/l con empagliflozina 25 mg una vez al día, respectivamente. La exposición sistémica de la empagliflozina se incrementó de manera proporcional a la dosis. Los parámetros farmacocinéticos de las dosis únicas de empagliflozina fueron similares a aquellos del estado de equilibrio dinámico, lo que sugiere una farmacocinética lineal en lo que al tiempo se refiere. No hubo ninguna diferencia clínicamente relevante en la farmacocinética de empagliflozina entre los voluntarios sanos y los pacientes con diabetes mellitus tipo 2.
La farmacocinética de una dosis de 5 mg de empagliflozina administrada dos veces al día y de dosis de 10 mg de empagliflozina administrada una vez al día se comparó en sujetos sanos. La exposición total (AUCss) de empagliflozina a lo largo de un período de 24 horas con dosis de 5 mg administradas dos veces al día fue similar a la observada en el caso del régimen de dosis de 10 mg administrada una vez al día. Tal como se esperaba, el régimen de dosis de empagliflozina 5 mg administradas dos veces al día en comparación con el régimen de dosis de empagliflozina de 10 mg administrada una vez al día condujo a valores más bajos de Cmax y a valores más altos de concentración plasmática valle (Cmin) de empagliflozina.
La administración de 25 mg de empagliflozina tras la ingesta de una comida de alto contenido graso y alto contenido calórico condujo a un ligero descenso de la exposición; el AUC se redujo en aproximadamente un 16% y la Cmax se redujo en aproximadamente un 37%, en comparación con los valores preprandiales registrados en ayunas. Este efecto de la ingesta de alimentos que se observó sobre la farmacocinética de la empagliflozina no se consideró clínicamente relevante, con lo cual la empagliflozina puede tomarse con las comidas o alejado de ellas.
Distribución: El volumen de distribución aparente en estado de equilibrio dinámico se estimó en un valor de 73,8 l, sobre la base de un análisis de farmacocinética poblacional. Tras la administración de una solución oral de [14C]-empagliflozina a sujetos sanos, el particionamiento en los glóbulos rojos fue de aproximadamente el 36,8% y el índice de unión a las proteínas plasmáticas fue del 86,2%.
Metabolismo: No se identificó ningún metabolito mayor de la empagliflozina en el plasma humano; los metabolitos más abundantes fueron tres conjugados glucurónidos (glucurónidos 2-O-, 3-O- y 6-O). La exposición sistémica de cada metabolito fue de menos del 10% del total del material relacionado con el fármaco. Los estudios in vitro sugirieron que la principal vía metabólica de la empagliflozina en los seres humanos es la glucuronidación a través de las uridina 5›-difosfo-glucuronosiltransferasas, UGT1A3, UGT1A8, UGT1A9 y UGT2B7.
Eliminación: La vida media de eliminación terminal aparente de la empagliflozina se estimó en un valor de 12,4 h, y la depuración oral aparente fue de 10,6 l/h sobre la base del análisis de farmacocinética poblacional. Las variabilidades intersujeto y residuales de la depuración oral de la empagliflozina fueron del 39,1% y del 35,8%, respectivamente. Con un régimen de una toma diaria, las concentraciones plasmáticas en estado de equilibrio dinámico de la empagliflozina se alcanzaron para la quinta dosis. En concordancia con su vida media, en el estado de equilibrio dinámico se observó una acumulación de hasta un 22%, con respecto al AUC plasmático. Tras la administración de una solución oral de [14C]-empagliflozina a sujetos sanos, aproximadamente el 95,6% de la radioactividad relacionada con el fármaco fue eliminada en las heces (41,2%) o en la orina (54,4%). La mayor parte de la radioactividad relacionada con el fármaco recuperada en las heces correspondía al fármaco original inalterado y aproximadamente la mitad de la radioactividad relacionada con el fármaco excretada en la orina era fármaco original inalterado.
Poblaciones específicas:
Insuficiencia renal: En los pacientes con insuficiencia renal leve (TFGe: 60 a <90 ml/min/1,73 m2), moderada (TFGe: 30 a <60 ml/min/1,73 m2), grave (TFGe: <30 ml/min/1,73 m2) y en los pacientes con falla renal/ ESRD (enfermedad renal en estadio final), el valor de AUC de la empagliflozina se incrementó aproximadamente un 18%, un 20%, un 66% y un 48%, respectivamente, en comparación con los sujetos con función renal normal. Los niveles plasmáticos pico de empagliflozina fueron similares en los sujetos con insuficiencia renal moderada y en los sujetos con falla renal/ESRD en comparación con los pacientes con función renal normal. Los niveles plasmáticos pico de empagliflozina fueron aproximadamente un 20% más altos en los sujetos con insuficiencia renal leve y grave en comparación con los sujetos con función renal normal. En concordancia con el estudio de fase I, el análisis de farmacocinética poblacional indicó que la depuración oral aparente de la empagliflozina se redujo con un descenso en la TFGe que condujo a un incremento en la exposición al fármaco. No se recomienda ningún ajuste de la posología en los pacientes con insuficiencia renal sobre la base de los datos de farmacocinética observados.
Insuficiencia hepática: En los sujetos con insuficiencia hepática leve, moderada y grave de acuerdo con la clasificación de Child-Pugh, el AUC de la empagliflozina se incrementó aproximadamente un 23%, un 47% y un 75% y la Cmax se incrementó aproximadamente un 4%, un 23% y un 48 %, respectivamente, en comparación con los sujetos con función hepática normal. No se recomienda ningún ajuste de la posología en los pacientes con insuficiencia hepática sobre la base de los datos de farmacocinética observados.
Índice de masa corporal (IMC): No se requiere ningún ajuste de la posología en función del IMC. El índice de masa corporal no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de la empagliflozina en función de lo determinado mediante el análisis de farmacocinética poblacional.
Género: No se requiere ningún ajuste de la posología en función del género. El género no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de la empagliflozina en función de lo determinado mediante el análisis de farmacocinética poblacional.
Raza: No se requiere ningún ajuste de la posología en función de la raza. Sobre la base del análisis de farmacocinética poblacional, se estimó que el AUC fue un 13,5% más alto en los pacientes asiáticos con un IMC de 25 kg/m2 en comparación con los pacientes de raza no asiática con un IMC de 25 kg/m2.
Pacientes geriátricos: La edad no tuvo ninguna repercusión clínicamente significativa sobre la farmacocinética de la empagliflozina en función de lo determinado mediante el análisis de farmacocinética poblacional.
Pacientes pediátricos: No se han realizado estudios para caracterizar la farmacocinética de la empagliflozina en pacientes pediátricos.
Metformina:
Absorción: Tras una dosis oral de metformina, el Tmax se alcanza en 2,5 horas. La biodisponibilidad absoluta de un comprimido de 500 mg o de 850 mg de clorhidrato de metformina es de aproximadamente un 50-60% en los sujetos sanos. Luego de una dosis oral, la fracción no absorbida recuperada en las heces fue del 20-30%.
Tras la administración oral, la absorción del clorhidrato de metformina es saturable e incompleta. Se presume que la farmacocinética de la absorción del clorhidrato de metformina es no lineal.
Con las dosis y los regímenes posológicos recomendados del clorhidrato de metformina, las concentraciones plasmáticas en estado de equilibrio dinámico se alcanzan dentro de un lapso de 24 a 48 horas y, por lo general, son inferiores a 1 μg/ml.
En los estudios clínicos comparativos, los niveles plasmáticos máximos (Cmax) del clorhidrato de metformina no superaron los 5 μg/ml, ni siquiera con las dosis máximas.
La ingesta de alimentos reduce el grado de absorción y demora ligeramente la absorción del clorhidrato de metformina. Luego de la administración de una dosis de 850 mg, se observó una concentración plasmática pico un 40% más baja, un descenso del 25% en el AUC y una prolongación de 35 minutos en el tiempo hasta la concentración plasmática pico. Se desconoce la relevancia clínica de estos descensos.
Distribución: El grado de unión a las proteínas plasmáticas es insignificante. El clorhidrato de metformina experimenta partición hacia los eritrocitos. El valor pico en sangre es menor que el pico plasmático y aparece aproximadamente en el mismo tiempo. Es muy probable que los glóbulos rojos representen un compartimiento de distribución secundario. El volumen medio de distribución (Vd) se ubicó en el rango de 63 a 276 l.
Metabolismo: El clorhidrato de metformina se excreta principalmente inalterado en la orina. No se han identificado metabolitos en los seres humanos.
Eliminación: La depuración renal del clorhidrato de metformina es >400 ml/min, lo que indica que el clorhidrato de metformina se elimina mediante filtración glomerular y secreción tubular. Luego de una dosis oral, la vida media de eliminación terminal aparente es de aproximadamente 6,5 horas. En presencia de un deterioro de la función renal, la depuración renal se reduce en forma proporcional a la disminución de la depuración de creatinina y, por lo tanto, la vida media de eliminación se prolonga, lo que conduce a niveles incrementados de clorhidrato de metformina en el plasma.
Poblaciones especiales:
Insuficiencia renal: Los datos disponibles de sujetos con insuficiencia renal moderada son escasos y no pudo hacerse una estimación confiable de la exposición sistémica a la metformina en este subgrupo en comparación con sujetos con función renal normal. Por ende, la dosis deberá adaptarse según consideraciones de eficacia clínica/tolerabilidad (véase la sección Posología y administración).
Pacientes pediátricos: Estudio de dosis únicas: Tras la administración de dosis únicas de metformina de 500 mg, los pacientes pediátricos han evidenciado un perfil farmacocinético similar al observado en adultos sanos.
Estudio de dosis múltiples: Tras la administración de dosis repetidas de 500 mg dos veces al día durante 7 días en pacientes pediátricos, la concentración plasmática pico (Cmax) y la exposición sistémica (AUC0-t) fueron aproximadamente un 33% y un 40% más bajas, respectivamente, en comparación con adultos diabéticos que recibieron dosis repetidas de 500 mg dos veces al día durante 14 días. Este aspecto tiene una relevancia clínica limitada, puesto que la dosis se ajusta en forma individual para cada caso en particular en función del control glucémico logrado.
CONTRAINDICACIONES:
• Hipersensibilidad a los principios activos empagliflozina y/o metformina, o a cualquiera de los excipientes (véase Composición).
• Cualquier tipo de acidosis metabólica aguda (como acidosis láctica, cetoacidosis diabética).
• Precoma diabético.
• Insuficiencia renal o disfunción renal (depuración de creatinina < 60 ml/min).
• Cuadros agudos con el potencial de alterar la función renal, como: deshidratación, infección grave, shock, administración intravascular de medios de contraste yodados (véase la sección Advertencias y precauciones especiales).
• Afección que pueda provocar hipoxia tisular (especialmente afección aguda o empeoramiento de afección crónica), como: insuficiencia cardíaca descompensada, insuficiencia respiratoria, infarto de miocardio reciente, shock.
• Insuficiencia hepática, intoxicación alcohólica aguda, alcoholismo (véase la sección Interacciones).
• En el caso de trastornos hereditarios raros que puedan ser incompatibles con alguno de los excipientes del producto, el uso de este producto está contraindicado.
• Menores de 18 años.
EFECTOS SECUNDARIOS: Un total de 12245 pacientes con diabetes tipo 2 fueron tratados en el marco de estudios clínicos para evaluar la seguridad de la combinación de empagliflozina más metformina, de los cuales 8199 fueron tratados con la combinación de empagliflozina más metformina, ya sea sola o sumada a una sulfonilurea, a pioglitazona, a inhibidores de la DPP4 o a insulina. En estos estudios, 2910 pacientes recibieron tratamiento con empagliflozina 10 mg más metformina y 3699 pacientes recibieron tratamiento con empagliflozina 25 mg más metformina durante al menos 24 semanas y 2151 o 2807 pacientes durante un mínimo de 76 semanas.
El perfil de seguridad global de la empagliflozina más metformina en los pacientes reclutados en el estudio EMPA-REG OUTCOME® fue similar al perfil de seguridad previamente conocido.
Los estudios de diseño doble ciego, comparativos con placebo de 18 a 24 semanas de exposición incluyeron 3456 pacientes, de los cuales 1271 fueron tratados con empagliflozina 10 mg más metformina y 1259 fueron tratados con empagliflozina 25 mg más metformina.
El evento adverso informado con mayor frecuencia en los estudios clínicos fue la hipoglucemia, el cual dependió del tipo de tratamiento de base utilizado en los respectivos estudios (véase la descripción de los Efectos secundarios seleccionados).
No se identificó ningún efecto secundario adicional en los estudios clínicos realizados con empagliflozina más metformina en comparación con los efectos secundarios de los componentes individuales.
|
Tabla 2. Efectos secundarios informados en pacientes que recibieron monoterapia de empagliflozina o tratamiento combinado de empagliflozina y metformina en estudios doble ciego, comparativos con placebo de hasta 24 semanas de duración (independientemente de la relación causal informada por el investigador), y efectos secundarios derivados de la experiencia posterior a la comercialización con monoterapia de empagliflozina o tratamiento combinado de empagliflozina y metformina, clasificados por sistema y órgano (SOC) del MedDRA y por término preferente del MedDRA. |
|
|
Empagliflozina y metformina |
|
|
Clasificación por sistema y órgano |
Efectos secundarios |
|
Infecciones e infestaciones |
Moniliasis vaginal, vulvovaginitis, balanitis y otras infecciones genitales1,2 Infección de las vías urinarias1,2 (incluso pielonefritis y urosepsis)6 Fascitis necrotizante del perineo (gangrena de Fournier)2,6 |
|
Trastornos gastrointestinales5 |
Náuseas3 Vómitos3 Diarrea3 Dolor abdominal3 Pérdida de apetito3 |
|
Trastornos del metabolismo y de la nutrición |
Hipoglucemia (cuando se usa con una sulfonilurea o con insulina)1 Acidosis láctica3 Disminución de la absorción de vitamina B123,4 Cetoacidosis6 |
|
Trastornos hepatobiliares |
Valores anómalos en las pruebas de función hepática3, Hepatitis3 |
|
Trastornos del sistema nervioso |
Alteración del sentido del gusto3 |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito2,3 Reacciones alérgicas cutáneas (p. ej. exantema6, urticaria3,6, eritema3) |
|
Trastornos vasculares |
Depleción de volumen2 |
|
Trastornos renales y urinarios |
Aumento de la micción2 Disuria2 |
|
Trastornos generales y condiciones en el lugar de la administración |
Sed2 |
|
Investigaciones |
Disminución de la tasa de filtración glomerular1 Aumento de la creatinina en sangre1 Aumento del hematocrito2,7 Aumento de lípidos en suero2,7 |
|
1 Véanse las subsecciones siguientes para obtener información adicional. 2 Efectos secundarios identificados de la monoterapia de empagliflozina. 3 Efectos secundarios identificados, sobre la base del SmPC de metformina para la UE 4 El tratamiento prolongado con metformina se ha asociado con una disminución de la absorción de vitamina B12, lo cual podría dar lugar a una deficiencia de vitamina B12 clínicamente significativa (p. ej., anemia megaloblástica). 5 Los síntomas gastrointestinales como las náuseas, los vómitos, la diarrea, el dolor abdominal y la falta de apetito se producen con la mayor frecuencia durante el inicio del tratamiento y se resuelven espontáneamente en la mayoría de los casos. 6 Efectos secundarios identificados a partir de la experiencia posterior a la comercialización 7 Véase la sección de estudios clínicos para información adicional |
|
Descripción de los efectos secundarios seleccionados: Las frecuencias que se indican a continuación se calcularon para los efectos secundarios independientemente de la causalidad.
Hipoglucemia: La frecuencia de la hipoglucemia dependió del tratamiento de base utilizado en los respectivos estudios y fue similar a la observada con placebo cuando la empagliflozina se administró como tratamiento complementario de un régimen de metformina y como tratamiento complementario de un régimen de pioglitazona +/- metformina, y como tratamiento complementario de un régimen de linagliptina + metformina. La frecuencia de pacientes con hipoglucemia se incrementó en pacientes tratados con empagliflozina en comparación con el placebo cuando se la administró como tratamiento complementario de un régimen de metformina más sulfonilurea, y como tratamiento complementario de un régimen de insulina +/- metformina y +/- sulfonilurea (véase la sección Posología y Administración; véase la Tabla 3 a continuación).
Hipoglucemia grave (eventos que requieren asistencia): La frecuencia general de pacientes con eventos de hipoglucemia grave fue baja (<1%) y similar para empagliflozina y para placebo con un tratamiento de base de metformina. La frecuencia de hipoglucemia grave dependió del tratamiento de base utilizado en los respectivos estudios (véase la sección Posología y Administración; véase la Tabla 3 a continuación).
|
Tabla 3. Frecuencia de pacientes con eventos hipoglucémicos confirmados por estudio e indicación (1245.19, 1245.23(met), 1245.23(met+SU), 1245.33, 1245.49, 1276.1, 1276.10, 1275.9 y 1245.25 - TS1) |
|||
|
Grupo de tratamiento |
Placebo |
Empagliflozina 10 mg |
Empagliflozina 25 mg |
|
En combinación con metformina (1245.23 (met)) (24 semanas) |
|||
|
N |
206 |
217 |
214 |
|
Total casos confirmados (%) |
0,5% |
1,8% |
1,4% |
|
Grave (%) |
0% |
0% |
0% |
|
En combinación con metformina + Sulfonilurea (1245.23 (met + SU)) (24 semanas) |
|||
|
N |
225 |
224 |
217 |
|
Total casos confirmados (%) |
8,4% |
16,1% |
11,5% |
|
Grave (%) |
0% |
0% |
0% |
|
En combinación con Pioglitazona +/- Metformina (1245.19) (24 semanas) |
|||
|
N |
165 |
165 |
168 |
|
Total casos confirmados (%) |
1,8% |
1,2% |
2,4% |
|
Grave (%) |
0% |
0% |
0% |
|
En combinación con insulina basal +/- Metformina +/- Sulfonilurea (1245.33) (18 semanas2/ 78 semanas) |
|||
|
N |
170 |
169 |
155 |
|
Total casos confirmados (%) |
20,6% / 35,3% |
19,5% / 36,1% |
28,4% / 36,1% |
|
Grave (%) |
0% / 0% |
0% / 0% |
1,3% / 1,3% |
|
En combinación con insulina MDI +/-Metformina (1245.49) (18 semanas2/ 52 semanas) |
|||
|
N |
188 |
186 |
189 |
|
Total casos confirmados (%) |
37,2% / 58,0% |
39,8% / 51,1% |
41,3% / 57,7% |
|
Grave (%) |
1,6% / 1,6% |
1,6% / 1,6% |
0,5% / 0,5% |
|
Empagliflozina dos veces al día versus una vez al día como tratamiento complementario de metformina (1276.10) (16 semanas) |
|||
|
Placebo |
Empa 10 mg |
Empa 25 mg |
|
|
N |
107 |
439 |
437 |
|
Total casos confirmados (%) |
0,9% |
0,5% |
0,2% |
|
Grave (%) |
0% |
0% |
0% |
|
En combinación con metformina en pacientes sin tratamiento previo (1276.13) (24 semanas) |
|||
|
Met 500/1000 mg dos veces al día |
Empa 10/25 mg una vez al día |
Empa (5/12,5 mg) + Met (500/1000 mg) dos veces al día |
|
|
N |
341 |
339 |
680 |
|
Total casos confirmados (%) |
0,6% |
0,6% |
1,0% |
|
Grave (%) |
0% |
0% |
0% |
|
En combinación con metformina y linagliptina (1275.9) (24 semanas)3 |
|||
|
N |
110 |
112 |
110 |
|
Total casos confirmados (%) |
0,9% |
0,0% |
2,7% |
|
Grave (%) |
0% |
0% |
0,9% |
|
Estudio EMPA REG OUTCOME (1245.25) |
|||
|
Placebo |
Empa 10 mg |
Empa 25 mg |
|
|
N |
2333 |
2345 |
2342 |
|
Total casos confirmados (%) |
27,9% |
28% |
27,6% |
|
Grave (%) |
1,5% |
1,4% |
1,3% |
|
Confirmado: glucosa en sangre ≤70 ml/dl o requirió asistencia Grave: requirió asistencia 1 A saber, pacientes que recibieron al menos una dosis del medicamento en estudio 2 La dosis de insulina como medicación de base debía ser estable durante las primeras 18 semanas 3 Ocho grupos de tratamiento: 4 tratamientos combinados de empagliflozina (5 mg o 12,5 mg dos veces al día) y metformina (500 o 1000 mg dos veces al día) y tratamiento con los componentes individuales de empagliflozina (10 mg o 25 mg una vez al día) o metformina (500 mg o 1000 mg dos veces al día). 3 Consistió en una combinación a dosis fija de empagliflozina con linagliptina 5 mg, con un tratamiento de base con metformina (véase también la sección Estudios Clínicos). Fuente de datos: 1245.19 [U12-1516, Tabla 15.3.2.3: 3], 1245.23 [U12-1518, Tablas 15.1.3.2.3: 3 y 15.2.3.2.3: 3], 1245.33 [U12-3817, Tablas 15.3.2.3: 3 y 15.4.5: 3], 1245.49 [U13-2122, Tablas 15.3.2.4: 3 y 15.3.2.5: 3], 1276.1 [c02661969, Tabla 15.3.1.3:4]; 1276.10 [c02092828-02, Tabla 15.3.2.3:3], 1275.9 [c02820144-01 Tabla 15.4.2: 12], 1245.25 [c02695839-01, Tabla 15.3.1.4: 4]. |
|||
Infección de las vías urinarias: La frecuencia general de eventos adversos de infección de las vías urinarias fue más alta en los pacientes tratados con empagliflozina 10 mg más metformina (8,8%) en comparación con empagliflozina 25 mg más metformina (6,6%) o placebo más metformina (7,8%). De manera similar a lo observado en el caso del placebo, el evento de infección de las vías urinarias fue informado con mayor frecuencia para la combinación de empagliflozina más metformina en los pacientes con antecedentes de infecciones urinarias crónicas o recurrentes. La intensidad de las infecciones de las vías urinarias fue similar a la observada con placebo. Los eventos de infecciones urinarias se informaron con mayor frecuencia en relación con el régimen de empagliflozina 10 mg más metformina, en comparación con placebo, en las pacientes de sexo femenino, pero no así en el caso de empagliflozina 25 mg más metformina. Las frecuencias de infecciones de las vías urinarias fueron bajas en el caso de los pacientes de sexo masculino y estuvieron equilibradas entre los grupos de tratamiento.
Moniliasis vaginal, vulvovaginitis, balanitis y otras infecciones genitales: Los eventos de moniliasis vaginal, vulvovaginitis, balanitis y otras infecciones genitales fueron informados con mayor frecuencia para empagliflozina 10 mg más metformina (4,0%) y para empagliflozina 25 mg más metformina (3,9%) en comparación con placebo más metformina (1,3%), y fueron informados con mayor frecuencia para empagliflozina más metformina en comparación con el placebo en las pacientes de sexo femenino. La diferencia en términos de frecuencia fue menos pronunciada en los pacientes de sexo masculino. Las infecciones genitales fueron de intensidad leve y moderada, y en ninguno de los casos fueron de intensidad grave.
Aumento de la micción: Tal como era de esperar por su mecanismo de acción, se observó un aumento de la micción (según lo evaluado por la búsqueda por término preferente, que incluye polaquiuria, poliuria y nicturia) con mayor frecuencia en los pacientes tratados con empagliflozina 10 mg más metformina (3,0%) y empagliflozina 25 mg más metformina (2,9%) en comparación con aquellos que recibieron placebo más metformina (1,4%). El aumento de la micción fue mayormente de intensidad leve o moderada. La frecuencia de la nicturia informada fue comparable entre el placebo y empagliflozina, ambos con un régimen de base de metformina (<1%).
Depleción de volumen: La frecuencia general de depleción de volumen (lo que incluye los términos predefinidos de descenso de la presión arterial (ambulatoria), descenso de la presión arterial sistólica, deshidratación, hipotensión, hipovolemia, hipotensión ortostática y síncope) fue baja y comparable a la observada en el caso de placebo (empagliflozina 10 mg más metformina (0,6%), empagliflozina 25 mg más metformina (0,3%) y placebo más metformina (0,1%). El efecto de la empagliflozina sobre la excreción urinaria de la glucosa está asociado con un mecanismo de diuresis osmótica, el cual podría afectar la hidratación en los pacientes de 75 años de edad o más. En los pacientes de ≥75 años de edad, los eventos de depleción de volumen han sido informados en un único paciente tratado con empagliflozina 25 mg más metformina.
Aumento de la creatinina en sangre y disminución de la tasa de filtración glomerular: La frecuencia global de pacientes con aumento de la creatinina en sangre y disminución de la tasa de filtración glomerular fue similar entre empagliflozina y placebo como tratamiento complementario de un régimen con metformina (aumento de la creatinina en sangre: empagliflozina 10 mg 0,5%, empagliflozina 25 mg 0,1%, placebo 0,4%; disminución de la tasa de filtración glomerular: empagliflozina 10 mg 0,1%, empagliflozina 25 mg 0%, placebo 0,2%).
En estos estudios doble ciego, controlados con placebo de 24 semanas de duración, se observaron aumentos transitorios iniciales en los niveles de creatinina (cambio medio del nivel basal luego de 12 semanas: empagliflozina 10 mg 0,02 mg/dl, empagliflozina 25 mg 0,02 mg/dl) y disminuciones transitorias iniciales en las tasas de filtración glomerular estimadas (cambio medio del nivel basal luego de 12 semanas: empagliflozina 10 mg -1,46 ml/min/1,73 m2, empagliflozina 25 mg -2,05 ml/min/1,73 m2). En los estudios a largo plazo, estos cambios, en general, fueron reversibles durante el tratamiento continuo o luego de la interrupción del fármaco (véase la figura 6 en la Sección Estudios Clínicos para más información sobre el curso de TFGe en el estudio EMPA-REG outcome®)
INTERACCIONES: Empagliflozina: Interacciones farmacodinámicas: Diuréticos: El efecto de la empagliflozina puede sumarse al efecto diurético de los diuréticos tiazídicos y los diuréticos del asa, y puede incrementar el riesgo de deshidratación e hipotensión.
Insulina y secretagogos de la insulina: La insulina y los secretagogos de la insulina, como las sulfonilureas, pueden aumentar el riesgo de hipoglucemia. Por ende, puede requerirse una dosis menor de insulina o de un secretagogo de la insulina para reducir el riesgo de hipoglucemia cuando se los utiliza en combinación con la empagliflozina (véanse las secciones Posología y Administración y Efectos secundarios).
Interferencia con el Ensayo de 1,5-anhidroglucitol (1,5-AG): No se recomienda el monitoreo del control glucémico con el ensayo 1,5-AG, dado que las mediciones de 1,5-AG no son fiables para la evaluación del control glucémico en pacientes que reciben inhibidores de SGLT-2. Se deben utilizar métodos alternativos para el monitoreo del control glucémico.
Interacciones farmacocinéticas:
Evaluación in vitro de las interacciones medicamentosas: La empagliflozina no inhibe, inactiva ni induce las isoformas del CYP450. Los datos obtenidos in vitro sugieren que la principal vía metabólica de la empagliflozina en los seres humanos es su glucuronidación a través de las uridina 5’-difosfo-glucuronosiltransferasas UGT1A3, UGT1A8, UGT1A9 y UGT2B7. La empagliflozina no inhibe la UGT1A1, la UGT1A3, la UGT1A8, la UGT1A9 o la UGT2B7. En las dosis terapéuticas, el potencial de que empagliflozina inactive o inhiba de manera reversible las principales isoformas del CYP450 o la UGT1A1 es remota. Por lo tanto, se considera improbable que se produzcan interacciones medicamentosas de las principales isoformas del CYP450 y de la UGT con la empagliflozina y los sustratos de estas enzimas administrados en forma concomitante.
La empagliflozina es un sustrato de la glucoproteína P (P-gp) y de la proteína de resistencia del cáncer de mama (BCRP), pero no inhibe estos transportadores de eflujo en las dosis terapéuticas. Con base en estudios in vitro, se considera improbable que la empagliflozina tenga alguna interacción con los fármacos que son sustratos de la P-gp. La empagliflozina es un sustrato de los transportadores de captación humanos OAT3, OATP1B1 y OATP1B3, pero no de OAT1 y OCT2.
La empagliflozina no inhibe ninguno de estos transportadores de captación humanos en las concentraciones plasmáticas clínicamente relevantes y, por lo tanto, se considera improbable que se produzcan interacciones medicamentosas con los sustratos de estos transportadores de entrada.
Evaluación in vivo de las interacciones medicamentosas: No se observaron interacciones farmacocinéticas clínicamente significativas cuando la empagliflozina se coadministró junto con otros medicamentos de uso común. Sobre la base de los estudios de farmacocinética, no es necesario ningún ajuste de la dosis de la empagliflozina cuando este fármaco se coadministra con medicamentos comúnmente prescriptos.
La farmacocinética de la empagliflozina fue similar con y sin la coadministración de glimepirida, pioglitazona, sitagliptina, linagliptina, warfarina, verapamilo, ramipril, simvastatina, en voluntarios sanos y con o sin la coadministración de torasemida e hidroclorotiazida en pacientes con DMT2. Se observó un incremento de la exposición total (AUC) de la empagliflozina luego de la coadministración con gemfibrozil (59%), rifampicina (35%) o probenecid (53%). Estos cambios no fueron considerados clínicamente significativos.
La empagliflozina no tuvo ningún efecto clínicamente relevante en la farmacocinética de la glimepirida, la pioglitazona, la sitagliptina, la linagliptina, la warfarina, la digoxina, el ramipril, la simvastatina, la hidroclorotiazida, la torasemida ni los anticonceptivos orales cuando se coadministró en voluntarios sanos.
Metformina:
Uso concomitante no recomendado: Alcohol: La intoxicación alcohólica está asociada con un mayor riesgo de acidosis láctica, particularmente en presencia de factores como ayuno, malnutrición o insuficiencia hepática.
Medios de contraste yodados: La administración de JARDIANCE DUO® debe interrumpirse con anterioridad al procedimiento de diagnóstico por imágenes, o al momento de su realización, y deben dejarse transcurrir como mínimo 48 horas antes de reanudarlo, siempre que se haya reevaluado la función renal y comprobado que es estable (véanse las secciones Posología y Advertencias y precauciones especiales).
Combinaciones que requieren precauciones de uso: Algunos medicamentos pueden afectar de forma adversa la función renal, lo que puede incrementar el riesgo de acidosis láctica, p. ej., los AINEs, incluidos los inhibidores selectivos de la ciclooxigenasa (COX) II, los inhibidores de la ECA, los antagonistas del receptor de la angiotensina II y los diuréticos, en especial, los diuréticos del asa. Cuando se inicie un tratamiento con estos productos o se usen en combinación con metformina, es necesario un monitoreo estrecho de la función renal.
Transportadores de cationes orgánicos (OCT):
La metformina es un sustrato de ambos transportadores OCT1 y OCT2. La coadministración de metformina con: Los inhibidores de OCT1 (como el verapamilo) pueden reducir la eficacia de la metformina.
Los inductores de OCT1 (como la rifampicina) pueden aumentar la absorción gastrointestinal y la eficacia de la metformina.
Los inhibidores de OCT2 (como cimetidina, dolutegravir, ranolazina, trimetoprima, vandetanib, isavuconazol) pueden disminuir la eliminación renal de metformina y, por lo tanto, conducir a un aumento de la concentración plasmática de metformina.
Los inhibidores de OCT1 y OCT2 (como crizotinib, olaparib) pueden alterar la eficacia y la eliminación renal de la metformina.
Por lo tanto, se recomienda precaución, especialmente en pacientes con insuficiencia renal, cuando estos fármacos son coadministrados con metformina, ya que la concentración plasmática de metformina puede aumentar. Si es necesario, se puede considerar el ajuste de la dosis de metformina, ya que los inhibidores/inductores de los OCT pueden alterar la eficacia de la metformina.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES: Generalidades: JARDIANCE DUO® no debe utilizarse en pacientes con diabetes tipo 1.
Se ha observado un incremento en los casos de amputación de miembros inferiores principalmente de los dedos de los pies en ensayos clínicos a largo plazo con otro inhibidor de SGLT2. Se desconoce si esto constituye un efecto de clase. Al igual que para todos los pacientes diabéticos, es importante aconsejar a los pacientes acerca del cuidado rutinario preventivo de los pies.
Cetoacidosis diabética: Se han informado casos de cetoacidosis diabética (CAD), una afección seria potencialmente mortal que requiere hospitalización, en pacientes tratados con empagliflozina, incluidos casos mortales.
El riesgo de cetoacidosis diabética en pacientes tratados con inhibidores de SGLT2 debe considerarse ante la presencia de sintomatología inespecífica, como náuseas, vómitos, dolor abdominal, anorexia, sed excesiva, disnea, confusión, o cansancio o somnolencia inusual, incluso con niveles de glucemia menores de 250 mg/dL.
Los factores que deben tenerse en cuenta al inicio y durante el tratamiento con un inhibidor de SGLT2, comprenden situaciones que pueden predisponer a la presentación de cetoacidosis diabética, como deshidratación, restricción de ingesta calórica, reducción de peso, infecciones, cirugía, vómitos, reducción de la dosis de insulina, mal control de diabetes, o ingesta de alcohol; por lo cual estas situaciones deben tenerse en cuenta al prescribir un tratamiento con un inhibidor de SGLT2.
Si se sospecha el diagnóstico de cetoacidosis se debe suspender el tratamiento y realizar la determinación de cuerpos cetónicos.
Los pacientes que hayan tenido cetoacidosis durante el tratamiento con inhibidores de SGLT2 no se les debería reintroducir el tratamiento, a no ser que otros factores hayan sido claramente los precipitantes y éstos se hayan resuelto.
En caso de pacientes hospitalizados por cirugía mayor o enfermedad médica grave, el tratamiento con inhibidores de SGLT2 debe interrumpirse hasta que se resuelva la situación.
Estos medicamentos se encuentran exclusivamente indicados para el tratamiento de la diabetes mellitus tipo 2.
Acidosis láctica: La acidosis láctica, una complicación metabólica muy rara, pero seria, se produce con mayor frecuencia durante el empeoramiento agudo de la función renal, en caso de enfermedad cardiorrespiratoria o septicemia. La acumulación de metformina se produce durante el empeoramiento agudo de la función renal e incrementa el riesgo de acidosis láctica.
En caso de deshidratación (diarrea o vómitos intensos, fiebre o reducción de la ingesta de líquidos), la metformina se debe interrumpir de forma temporal y se recomienda contactar a un profesional sanitario.
El uso de medicamentos que puedan alterar de manera aguda la función renal (como antihipertensivos, diuréticos y AINEs) se debe iniciar con precaución en los pacientes tratados con metformina.
Otros factores de riesgo para la acidosis láctica son la ingesta excesiva de alcohol, la insuficiencia hepática, la diabetes mal controlada, la cetosis, el ayuno prolongado y cualquier trastorno asociado con hipoxia, así como el uso concomitante de medicamentos que puedan causar acidosis láctica (véase la sección Contraindicaciones e Interacciones).
Se debe informar a los pacientes y/o a las personas que los cuidan acerca del riesgo de acidosis láctica. La acidosis láctica se caracteriza por disnea acidótica, dolor abdominal, calambres musculares, astenia e hipotermia, seguidos de coma. En caso de que se sospeche de la presencia de síntomas, el paciente debe dejar de tomar metformina y buscar atención médica inmediata.
Los parámetros diagnósticos de laboratorio son descenso de los valores de pH sanguíneo (<7,35), aumento de los niveles plasmáticos de lactato (>5 mmol/l) y aumento del anión gap y del cociente lactato/piruvato.
Administración de un medio de contraste yodado: La administración intravascular de medios de contraste yodados puede provocar nefropatía inducida por el contraste, que puede ocasionar la acumulación de metformina y puede aumentar el riesgo de acidosis láctica. La administración de metformina se debe interrumpir con anterioridad al procedimiento de diagnóstico por imágenes, o al momento de su realización, y deben dejarse transcurrir como mínimo 48 horas antes de reanudarlo, siempre que se haya reevaluado la función renal y comprobado que es estable, véanse las secciones Posología e Interacciones.
Fascitis necrotizante del perineo (gangrena de Fournier): Riesgo de urosepsis, pielonefritis y gangrena de Fournier (fascitis necrotizante genital, perineal y perianal).
Se han informado casos posteriores a la comercialización de fascitis necrotizante del perineo (también denominada “gangrena de Fournier”), en hombres y mujeres con diabetes melitus tratados con inhibidores del SGLT2, como por ejemplo empagliflozina. Es una infección necrotizante rara pero seria y puede ser letal. Los casos serios han evolucionado con internación, intervenciones quirúrgicas múltiples y muerte.
Se debe descartar el diagnóstico de fascitis necrotizante en pacientes tratados con JARDIANCE DUO® que refieran dolor o molestias, eritema e inflamación en la zona genital o del perineo, fiebre, malestar general. En caso de que se sospeche dicho diagnóstico, la administración de Jardiance se debe discontinuar y se debe instituir un tratamiento de inmediato (inluso antibióticos de amplio espectro e intervención quirúrigca).
Función renal: Debido al mecanismo de acción, la eficacia de la empagliflozina depende de la función renal.
Se debe evaluar la TFG antes de iniciar el tratamiento y periódicamente a partir de entonces, véase la sección Posología. JARDIANCE DUO® está contraindicado en pacientes con TFG <60 ml/min/1.73 m2 y se debe interrumpir de forma temporal en presencia de trastornos que alteren la función renal, véase la sección Contraindicaciones.
Función cardíaca: Los pacientes con insuficiencia cardíaca tienen un mayor riesgo de padecer hipoxia e insuficiencia renal. En pacientes con insuficiencia cardíaca crónica estable, puede utilizarse JARDIANCE DUO® con un control periódico de la función cardíaca y la función renal.
En pacientes con insuficiencia cardíaca inestable y aguda, el uso de JARDIANCE DUO® está contraindicado debido al componente de la metformina (véase la sección Contraindicaciones).
Pacientes de edad avanzada: Los pacientes de 75 años de edad o más pueden tener un riesgo incrementado de depleción de volumen, por lo tanto, JARDIANCE DUO® debe prescribirse con precaución en estos pacientes (véase la sección Efectos secundarios). La experiencia terapéutica en pacientes de 85 años de edad o más es limitada. No se recomienda el inicio del tratamiento en dicha población.
En vista de que la metformina se excreta por vía renal y los pacientes de edad avanzada tienen una tendencia hacia un deterioro de la función renal, debe efectuarse un control periódico de la función renal en los pacientes de edad avanzada que reciban tratamiento con JARDIANCE DUO®.
Uso en pacientes con riesgo de depleción de volumen: Sobre la base del modo de acción de los inhibidores del SGLT-2, la diuresis osmótica que acompaña a la glucosuria terapéutica puede conducir a un ligero descenso de la presión arterial. Por lo tanto, debe tenerse precaución en los pacientes en los cuales un descenso en la presión arterial inducido por la empagliflozina podría suponer un riesgo, tal como pacientes con enfermedad cardiovascular conocida, pacientes en tratamiento con antihipertensivos con antecedentes de hipotensión o pacientes de 75 años de edad o más.
En el caso de patologías que pueden conducir a una pérdida de líquidos (p. ej., enfermedad gastrointestinal), se recomienda un monitoreo cuidadoso del estado de volumen (p. ej., examen físico, mediciones de presión arterial, pruebas de laboratorio, incluyendo nivel de hematocrito) y de los electrolitos en los pacientes que reciben empagliflozina. Debe considerarse la interrupción temporal del tratamiento con JARDIANCE DUO® hasta que se corrija la pérdida de líquidos.
Infecciones de las vías urinarias: En los estudios combinados, doble ciego, comparativos con placebo, de 18 a 24 semanas de duración, la frecuencia general de infecciones de las vías urinarias informada como un evento adverso fue más alta en los pacientes tratados con empagliflozina 10 mg más metformina en comparación con la observada en el caso de los pacientes tratados con placebo más metformina o empagliflozina 25 mg más metformina (véase la sección Efectos secundarios).
Se han informado casos posteriores a la comercialización de infecciones complicadas de las vías urinarias, incluso pielonefritis y urosepsis, en pacientes tratados con empagliflozina. Debe considerarse la interrupción temporal del tratamiento en los pacientes con infecciones complicadas de las vías urinarias.
Cirugía: JARDIANCE DUO® debe interrumpirse en el momento de la realización de una cirugía con anestesia general, raquídea o epidural. El tratamiento podrá reiniciarse luego de que haya transcurrido un mínimo de 48 horas desde la cirugía o la reanudación de la alimentación oral, y siempre que se haya reevaluado la función renal y comprobado que es estable.
Posibilidad de riesgo de amputación no traumática de miembros inferiores
POSOLOGÍA Y ADMINISTRACIÓN: Adultos con función renal normal (tasa de filtración glomerular estimada [TFGe] ≥ 90 ml/min): La posología debe individualizarse en función del régimen actual del paciente, la eficacia y la tolerabilidad. La dosis diaria máxima recomendada de JARDIANCE DUO® es de 25 mg de empagliflozina y 2000 mg de metformina.
• En los pacientes en los que no se logra un control adecuado con metformina sola o en combinación con otros productos, entre ellos insulina, la dosis inicial recomendada de JARDIANCE DUO® debe aportar empagliflozina en dosis de 5 mg administradas dos veces al día (dosis diaria total de 10 mg) y una dosis de metformina que sea similar a la que ya está recibiendo el paciente. En los pacientes que toleran una dosis diaria total de empagliflozina de 10 mg, la dosis se puede aumentar a una dosis diaria total de empagliflozina de 25 mg.
• Los pacientes que ya estén siendo tratados con empagliflozina deben continuar recibiendo la misma dosis diaria de empagliflozina.
• Los pacientes que pasen de un régimen de comprimidos separados de empagliflozina (dosis diaria total de 10 mg o 25 mg) y de metformina a un régimen de JARDIANCE DUO® deben recibir la misma dosis diaria de empagliflozina y de metformina que ya estén recibiendo, o la dosis de metformina lo más cercana posible que sea terapéuticamente adecuada.
Cuando JARDIANCE DUO® se usa en combinación con una sulfonilurea y/o con insulina, puede ser necesario el uso de una dosis menor de la sulfonilurea y/o de la insulina para reducir el riesgo de que se produzca un cuadro de hipoglucemia (véanse las secciones Interacciones y Efectos secundarios).
La dosis recomendada es un comprimido dos veces al día.
Para las diferentes dosis de metformina, JARDIANCE DUO® se encuentra disponible en las concentraciones de 5 mg de empagliflozina más 500 mg, 850 mg o 1000 mg de clorhidrato de metformina o bien 12,5 mg de empagliflozina más 500 mg, 850 mg o 1000 mg de clorhidrato de metformina.
JARDIANCE DUO® debe tomarse junto con las comidas para reducir los efectos indeseables gastrointestinales asociados con la metformina.
Insuficiencia renal: No se recomienda un ajuste de la dosis en pacientes con insuficiencia renal leve.
Se debe evaluar la TFG antes de iniciar el tratamiento con productos que contengan metformina y, al menos una vez al año a partir de entonces. En los pacientes expuestos a un mayor riesgo de progresión de la insuficiencia renal y en pacientes de edad avanzada, se debe evaluar la función renal con mayor frecuencia, p. ej., cada 3-6 meses.
Dosis omitidas: Si el paciente olvida una dosis, deberá tomarla tan pronto como lo recuerde. Sin embargo, no debe tomar una dosis doble en una misma toma. Si ese fuera el caso, deberá omitir la dosis olvidada.
Población pediátrica: No se recomienda el uso de JARDIANCE DUO® en niños y adolescentes menores de 18 años de edad, dada la falta de datos respecto de su seguridad y eficacia.
SOBREDOSIS: Durante los estudios clínicos controlados que se efectuaron en sujetos sanos, dosis únicas de hasta 800 mg de empagliflozina, equivalentes a 32 veces la dosis diaria máxima recomendada, fueron bien toleradas. No existe experiencia con dosis superiores a 800 mg en los seres humanos.
No se ha observado hipoglucemia con dosis de clorhidrato de metformina de hasta 85 g, si bien se han producido cuadros de acidosis láctica en dichas circunstancias. Una sobredosis de gran magnitud de clorhidrato de metformina o la presencia de riesgos concomitantes puede dar lugar a un cuadro de acidosis láctica. La acidosis láctica es una emergencia médica y debe ser tratada en el hospital.
Tratamiento: En el caso de una sobredosis, debe iniciarse el tratamiento de soporte que sea pertinente en función del estado clínico del paciente. El método más eficaz para eliminar del organismo el lactato y el clorhidrato de metformina es la hemodiálisis, en tanto que la remoción de la empagliflozina mediante hemodiálisis no ha sido estudiada.
TOXICOLOGÍA: Empagliflozina y metformina: Se llevaron a cabo estudios de toxicidad general en ratas de hasta 13 semanas de duración con la combinación de empagliflozina y metformina. En un estudio de la combinación de empagliflozina y metformina de 13 semanas de duración en ratas, la dosis (máxima) sin efectos adversos observados (NOAEL) se basó en la hipocloremia observada con exposiciones de aproximadamente 24 y 9 veces la exposición clínica basada en el AUC de empagliflozina asociada con las dosis de 10 y 25 mg, respectivamente.
Un estudio de desarrollo embriofetal realizado en ratas preñadas no indicó ningún efecto teratogénico atribuido a la coadministración de empagliflozina y metformina en exposiciones de aproximadamente 35 y 14 veces la exposición clínica de empagliflozina sobre la base del AUC asociada con las dosis de 10 y 25 mg, respectivamente, y 4 veces la exposición clínica basada en el AUC de metformina asociada con la dosis de 2000 mg. En el nivel de dosis de 600 mg/kg/día, asociado con niveles equivalentes a 8 veces la dosis recomendada máxima en los seres humanos (MRHD) de metformina en los seres humanos, se observó teratogenicidad de metformina.
Los datos que se brindan a continuación son hallazgos de estudios realizados con empagliflozina o metformina en forma individual.
Empagliflozina: En estudios de toxicidad general efectuados en roedores y en perros, se observaron signos de toxicidad con exposiciones superiores o iguales a 10 veces la dosis clínica de 25 mg. La mayor parte de la toxicidad observada fue concordante con una farmacología secundaria relacionada con la pérdida de glucosa urinaria, y comprendió el descenso del peso corporal y la reducción de la grasa corporal, un mayor consumo de alimentos, diarrea, deshidratación, descenso de los niveles séricos de glucosa y elevaciones en otros parámetros séricos que reflejan un aumento del metabolismo de las proteínas, gluconeogénesis y desequilibrios electrolíticos, cambios urinarios tales como poliuria y glucosuria, y cambios microscópicos en el riñón.
Carcinogenia: La empagliflozina no incrementó la incidencia de tumores en las ratas hembra en dosis de hasta la dosis más alta de 700 mg/kg/día, valor que corresponde a aproximadamente 72 y 182 veces la exposición clínica basada en el AUC asociada con las dosis de 25 mg y 10 mg, respectivamente. En las ratas macho, se observaron lesiones proliferativas vasculares benignas (hemangiomas) de los ganglios mesentéricos relacionadas con el tratamiento con dosis de 700 mg/kg/día, que corresponde a aproximadamente 42 y 105 veces la exposición clínica asociada con las dosis de 25 mg y 10 mg, respectivamente. Estos tumores son comunes en las ratas, y es improbable que sean de relevancia para los seres humanos. La empagliflozina no incrementó la incidencia de tumores en los ratones hembra en dosis de hasta 1000 mg/kg/día, valor que corresponde a aproximadamente 62 y 158 veces la exposición clínica asociada con las dosis de 25 mg y 10 mg, respectivamente. La empagliflozina indujo tumores renales en los ratones macho en dosis de 1000 mg/kg/día, valor que corresponde a aproximadamente 45 y 113 veces la exposición clínica asociada con las dosis de 25 mg y 10 mg, respectivamente. El modo de acción para estos tumores es dependiente de la predisposición natural de los ratones macho a las patologías renales y es una vía metabólica que no refleja la de los seres humanos. Los tumores renales observados en los ratones macho no se consideran relevantes para los seres humanos.
Genotoxicidad: La empagliflozina no es genotóxica.
Toxicidad para la reproducción: Los estudios preclínicos demuestran que la empagliflozina atraviesa la placenta durante la última fase de la gestación en un grado muy limitado, pero no indican efectos perjudiciales directos ni indirectos en lo que respecta al desarrollo embrionario temprano. La empagliflozina administrada durante el período de organogénesis no fue teratógena en dosis de hasta 300 mg/kg en las ratas ni en los conejos; dicho valor es aproximadamente 48 y 122 veces, o bien 128 y 325 veces, más alto que la exposición clínica de 25 mg y 10 mg, respectivamente, sobre la base de la exposición del AUC. Las dosis de empagliflozina que causaron toxicidad materna en las ratas también causaron malformaciones consistentes en huesos de extremidades curvados con exposiciones aproximadamente 155 y 393 veces más altas que la exposición clínica asociada con las dosis de 25 mg y 10 mg, respectivamente. Las dosis que causaron toxicidad materna en los conejos también ocasionaron un incremento en las pérdidas embriofetales en niveles de dosis aproximadamente 139 y 353 veces más altos que la exposición clínica asociada con las dosis de 25 mg y 10 mg, respectivamente.
En estudios de toxicidad pre- y posnatal realizados en ratas, se observó menor aumento de peso en las crías con exposiciones maternas equivalentes a aproximadamente 4 y 11 veces la dosis clínica asociada con las dosis de 25 mg y 10 mg, respectivamente.
En un estudio de toxicidad juvenil realizado en ratas, cuando la empagliflozina se administró desde el día 21 postnatal hasta el día 90 postnatal, se observó dilatación tubular y de la pelvis renal mínima a moderada, no adversa, en ratas jóvenes, solo en dosis de 100 mg/kg/día, lo cual se aproxima a 11 veces la dosis clínica máxima de 25 mg. Estos resultados no se observaron al cabo de un período de recuperación de 13 semanas sin uso de fármacos.
Metformina: Datos no clínicos no revelaron ningún peligro especial para los seres humanos sobre la base de los estudios convencionales de farmacología de seguridad, genotoxicidad y potencial carcinogénico. En un estudio de metformina sola de 2 semanas de duración y en estudios de empagliflozina/metformina de toxicidad de 2 y 13 semanas de duración en ratas, se observó toxicidad relacionada con la metformina en el corazón, el hígado, los riñones, las glándulas salivales, los ovarios, el aparato gastrointestinal y las glándulas suprarrenales con posologías asociadas con una exposición sistémica de 5 veces la MRHD o más.
La metformina no fue teratogénica en ratas en una dosis de 200 mg/kg/día asociada con una exposición sistémica de 4 veces la MRHD (2000 mg de metformina). Con dosis más elevadas (500 y 1000 mg/kg/día, asociadas con 11 y 23 veces la MRHD), se observó teratogenicidad de la metformina en las ratas lo cual se evidenció mayormente como un incremento de la cantidad de malformaciones esqueléticas.
PRESENTACIÓN: JARDIANCE® DUO 12,5 mg/ 850 mg, caja con blíster por 60 tabletas recubiertas (Reg. San. No. INVIMA 2016M-0017436).
JARDIANCE® DUO 12,5 mg/ 1000 mg, caja con blíster por 60 tabletas recubiertas (Reg. San. No. INVIMA 2016M-0017418).
¡Almacenar en un lugar seguro; fuera del alcance de los niños!
La información de seguridad del producto puede cambiar, consulte la información vigente en la Dirección Médica.
Teléfono: (601) 319 91 00, e-mail: medfora.co@boehringer-ingelheim.com
Carrera 11 No. 84A-09 Piso 5, Bogotá D.C. Colombia.
BOEHRINGER INGELHEIM S.A.
Versión- 13 Mod. del 19 de Octubre de 2018


