

KEYTRUDA
PEMBROLIZUMAB
Solución para infusión intravenosa
Vial(es), 4 mL,
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN:
Particularidades farmacéuticas:
Química: KEYTRUDA® (pembrolizumab) es un anticuerpo monoclonal humanizado selectivo diseñado para bloquear la interacción entre PD-1 y sus ligandos, PD-L1 y PD-L2. Pembrolizumab es una inmunoblogulina IgG4 kappa con un peso molecular aproximado de 149 kDa.
Principio activo: Pembrolizumab.
Componentes inactivos (Lista de excipientes):
L-histidina.
L-histidina hidrocloruro monohidrato.
Sacarosa.
Polisorbato 80.
Agua para inyección.
TOXICOLOGÍA ANIMAL:
Toxicidad crónica: Se evaluó la seguridad de pembrolizumab en un estudio de toxicidad de 1 mes y en un estudio de toxicidad de dosis repetida de 6 meses en monos Cynomolgus, administrando dosis IV de 6, 40 o 200 mg/kg una vez por semana en el estudio de 1 mes y una vez cada dos semanas en el estudio de 6 meses, seguido por un período libre de tratamiento de 4 meses. No se observaron hallazgos toxicológicamente importante y en ambos estudios, el nivel sin efecto adverso observable fue ≥200 mg/kg, que es 19 veces la exposición en los seres humanos a la dosis más alta clínicamente probada (10 mg/kg) y 94 veces la exposición en los seres humanos a la dosis recomendada (2 mg/kg).
Carcinogénesis: El potencial carcinogénico de pembrolizumab no ha sido evaluado en estudios en animales a largo plazo.
Mutagénesis: No se ha evaluado el potencial genotóxico de pembrolizumab.
Reproducción: No se han realizado estudios de reproducción animal con KEYTRUDA®. La función central de la vía PD-1/PD-L1 es preservar el embarazo, manteniendo la tolerancia inmunológica al feto. Se ha demostrado, en modelos murinos de embarazo, que el bloqueo de la señalización de PD-L1 altera la tolerancia al feto y resulta en un aumento de la pérdida fetal. Estos resultados indican un riesgo potencial de que la administración de KEYTRUDA® durante el embarazo podría causar daño fetal, incluyendo aumento de las tasas de aborto o muerte fetal.
Desarrollo: No se han realizado estudios de toxicidad sobre el desarrollo con pembrolizumab. No hubo efectos notables en los órganos reproductivos masculinos y femeninos en los monos basados en los estudios de toxicidad de dosis repetidas de 1 mes y 6 meses.
INDICACIONES Y USO:
Melanoma: KEYTRUDA® (pembrolizumab) está indicado para el tratamiento de pacientes con melanoma no resecable o metastásico.
Carcinoma de pulmón de células no pequeñas: KEYTRUDA® está indicado para el tratamiento de primera línea de pacientes con carcinoma de pulmón de células no pequeñas (NSCLC, por sus siglas en inglés) metastásico, cuyos tumores expresan PD-L1 con un ≥50% de puntuación de proporción de células tumorales (PPT), determinado por una prueba validada, sin aberraciones tumorales genómicas de EGFR o ALK.
KEYTRUDA® está indicado para el tratamiento de pacientes con NSCLC, cuyos tumores expresan PD-L1 con un ≥1% PPT, determinado mediante una prueba validada y que han recibido quimioterapia con platino. Los pacientes con aberraciones tumorales genómicas de EGFR o ALK deben haber recibido la terapia previa para estas aberraciones antes de recibir KEYTRUDA®.
USO EN POBLACIONES ESPECÍFICAS:
Embarazo: No existen datos sobre el uso de pembrolizumab en mujeres embarazadas. Tampoco se han realizado estudios de reproducción animal con pembrolizumab; sin embargo, se ha demostrado que el bloqueo de la señalización PD-L1, en modelos murinos de embarazo, altera la tolerancia al feto y resulta en un aumento de pérdidas fetales. Estos resultados indican un riesgo potencial, en base a su mecanismo de acción, que la administración de KEYTRUDA® durante el embarazo podría causar daño fetal, incluyendo aumento de las tasas de aborto o muerte fetal. Se sabe que la IgG4 humana (inmunoglobulina) cruza la barrera placentaria y el pembrolizumab es una IgG4; por tanto, el pembrolizumab tiene el potencial de ser transmitido de la madre al feto en desarrollo. KEYTRUDA® no se recomienda durante el embarazo, a menos que el beneficio clínico supere el riesgo potencial para el feto. Las mujeres en edad fértil deben utilizar anticoncepción eficaz durante el tratamiento con KEYTRUDA® y al menos durante 4 meses después de la última dosis de KEYTRUDA®.
Madres lactantes: Se desconoce si KEYTRUDA® es secretada en la leche humana. Debido a que muchos medicamentos se secretan en la leche humana, se debe tomar una decisión de descontinuar la lactancia o suspender KEYTRUDA®, teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio de KEYTRUDA® para la madre.
FARMACOCINÉTICA: La farmacocinética de pembrolizumab fue estudiada en 2188 pacientes con melanoma irresecable o metastásico, NSCLC, u otros carcinomas que recibieron dosis en el rango de 1 a 10 mg/kg cada 2 o 3 semanas.
Absorción: KEYTRUDA® se dosificó a través de la vía IV y por tanto es inmediata y completamente biodisponible.
Distribución: Consistente con una distribución extravascular limitada, el volumen de distribución de pembrolizumab, en estado de equilibrio, es pequeño (7.5 L; coeficiente de variación [CV]: 21%). Como era de esperarse para un anticuerpo, pembrolizumab no se une a las proteínas plasmáticas de una manera específica.
Metabolismo: El pembrolizumab es catabolizado a través de vías no específicas; el metabolismo no contribuye a su depuración.
Eliminación: La depuración sistémica de pembrolizumab es ~0.2 L/día (CV: 37%) y la vida media terminal (t½) es de ~26 días (CV: 39%).
La exposición a pembrolizumab expresada por la concentración máxima (Cmáx) o el área bajo la curva (AUC) de concentración plasmática-tiempo aumentó la dosis proporcionalmente dentro del rango de dosis para la eficacia. Después de dosis repetidas, se encontró que la depuración de pembrolizumab es independiente del tiempo y la acumulación sistémica fue aproximadamente 2.2 veces cuando se administró cada 3 semanas. Concentraciones de pembrolizumab cercanas al estado de equilibrio se alcanzaron a las 18 semanas; la mediana de la Cmín en la semana 18 es 22.8 mcg/mL, con dosis de 2 mg/kg cada 3 semanas.
Poblaciones especiales: En el análisis farmacocinético poblacional, se evaluaron los efectos de varias co-variables sobre la farmacocinética de pembrolizumab. La depuración de pembrolizumab aumentó con el aumento del peso corporal; las diferencias resultantes de la exposición se abordan adecuadamente con la administración sobre una base de mg/kg. Los siguientes factores no tuvieron ningún efecto clínicamente importante en la depuración de pembrolizumab: edad (rango 15-94 años), género, raza, insuficiencia renal leve o moderada, insuficiencia hepática leve y carga tumoral.
Insuficiencia renal: Se evaluó el efecto de la insuficiencia renal sobre la depuración de pembrolizumab mediante el análisis farmacocinético poblacional en pacientes con insuficiencia renal leve (GFR <90 y ≥60 mL/min/1.73 m2) o moderada (GFR <60 y ≥30 mL/min/1.73 m2) en comparación con los pacientes con función renal normal (GFR ≥90 mL/min/1.73 m2). No se encontraron diferencias clínicamente importantes en la depuración de pembrolizumab entre los pacientes con insuficiencia renal leve o moderada y los pacientes con función renal normal. KEYTRUDA® no ha sido estudiado en pacientes con insuficiencia renal severa (GFR <30 y ≥15 mL/min/1.73 m2). [Ver Dosis y administración]
Insuficiencia hepática: Se evaluó el efecto de la insuficiencia hepática sobre la depuración de pembrolizumab mediante el análisis farmacocinético poblacional en pacientes con insuficiencia hepática leve (bilirrubina total (BT) 1.0 a 1.5 veces el LSN o AST >LSN, como se define utilizando los criterios de disfunción hepática del National Cancer Institute) en comparación con pacientes con función hepática normal (BT y AST ≤LSN). No se encontraron diferencias clínicamente importantes en la depuración de pembrolizumab entre los pacientes con insuficiencia hepática leve y los pacientes con función hepática normal. KEYTRUDA® no ha sido estudiado en pacientes con insuficiencia hepática moderada (BT >1.5 a 3 veces el LSN y cualquier AST) o insuficiencia hepática severa (BT >3 veces el LSN y cualquier AST). [Ver Dosis y administración].
FARMACODINÁMICA: En la sangre periférica de pacientes que recibieron KEYTRUDA® 2 mg/kg cada 3 semanas o 10 mg/kg cada 2 semanas o 3 semanas, se observó un aumento del porcentaje de células T activadas (es decir, HLA-DR+) CD4+ y CD8+, después del tratamiento con todas las dosis y esquemas, sin aumento del número de linfocitos T circulantes.
FORMA FARMACÉUTICA: Solución transparente a ligeramente opalescente, incolora a ligeramente amarilla.
CONTRAINDICACIONES: KEYTRUDA® está contraindicado en pacientes con hipersensibilidad a pembrolizumab o a alguno de sus excipientes.
REACCIONES ADVERSAS:
Experiencia en los ensayos clínicos: La seguridad de KEYTRUDA® fue evaluada en 2799 pacientes en estudios controlados y no controlados. La duración promedio del tratamiento fue de 4.2 meses (rango 1 día a 30.4 meses) incluyendo 1153 pacientes tratados durante seis meses o más y 600 pacientes tratados durante un año o más. KEYTRUDA® fue descontinuado por reacciones adversas relacionadas con el tratamiento en el 5% de los pacientes. Los Eventos Adversos Serios (EAS) relacionados con el tratamiento reportados hasta 90 días después de la última dosis se presentaron en el 10% de los pacientes que recibieron KEYTRUDA®. De estos EAS relacionados con el tratamiento, los más comunes fueron neumonitis, colitis, diarrea y pirexia.
Reacciones adversas mediadas inmunológicamente [ver Advertencias y precauciones]: Las reacciones adversas mediadas inmunológicamente se presentan en base a 2799 pacientes con melanoma y NSCLC. El perfil de seguridad fue generalmente similar para los pacientes con melanoma y NSCLC. La Tabla 1 presenta la incidencia de las reacciones adversas mediadas inmunológicamente de acuerdo al Grado que se produjeron en pacientes que recibieron KEYTRUDA®.
|
Tabla 1: Reacción adversas mediadas inmunológicamente |
|||||
|
KEYTRUDA® 2 mg/kg cada 3 semanas o 10 mg/kg cada 2 o 3 semanas n=2117 |
|||||
|
Reacción adversa |
Todos los grados (%) |
Grado 2 (%) |
Grado 3 (%) |
Grado 4 (%) |
Grado 5 (%) |
|
Hipotiroidismo* |
8.5 |
6.2 |
0.1 |
0 |
0 |
|
Hipertiroidismo |
3.4 |
0.8 |
0.1 |
0 |
0 |
|
Neumonitis |
3.4 |
1.3 |
0.9 |
0.3 |
0.1 |
|
Colitis |
1.7 |
0.4 |
1.1 |
<0.1 |
0 |
|
Hepatitis |
0.7 |
0.1 |
0.4 |
<0.1 |
0 |
|
Hipofisitis |
0.6 |
0.2 |
0.3 |
<0.1 |
0 |
|
Nefritis |
0.3 |
0.1 |
0.1 |
<0.1 |
0 |
|
Diabetes Mellitus Tipo 1 |
0.2 |
<0.1 |
0.1 |
0.1 |
0 |
|
* En pacientes con Carcinoma de Cabeza y Cuello Escamoso Celular HNSCC (n=192) la incidencia de hipotiroidismo fue 14.1% (todos los grados) con 0.5% Grado 3. En pacientes con Linfoma Hodgkin clásico cHL (n=241) la incidencia de hipotiroidismo fue 12.9%; todos los casos fueron Grado 1 o 2. |
|||||
Endocrinopatías: La mediana de tiempo hasta la aparición de la hipofisitis fue de 3.7 meses (rango 1 día a 11.9 meses). La mediana de la duración fue de 4.7 meses (rango 8+ días a 12.7 ± meses). La hipofisitis condujo a discontinuación de KEYTRUDA® en 4 pacientes (0.1%). La hipofisitis fue resuelta en 7 pacientes. La mediana de tiempo hasta la aparición del hipertiroidismo fue de 1.4 meses (rango 1 día a 21.9 meses). La mediana de la duración fue de 2.1 meses (rango 3 días a 15.0+ meses). El hipertiroidismo provocó discontinuación de KEYTRUDA® en 2 pacientes (<0.1%). El hipertiroidismo se resolvió en 71 pacientes. La mediana de tiempo hasta la aparición del hipotiroidismo fue de 3.5 meses (rango 1 día a 18.9 meses). La mediana de la duración no se alcanzó (rango 2 días a 27.7+ meses). Un (<0.1%) paciente suspendió KEYTRUDA® debido a hipotiroidismo.
Neumonitis: La mediana de tiempo hasta la aparición de neumonitis fue de 3.3 meses (rango 2 días a 19.3 meses). La mediana de la duración fue de 1.5 meses (rango 1 día a 17.2+ meses). La neumonitis condujo a la discontinuación de KEYTRUDA® en 36 pacientes (1.3%). La neumonitis se resolvió en 55 pacientes.
Colitis: La mediana de tiempo hasta la aparición de colitis fue de 3.5 meses (rango 10 días a 16.2 meses). La mediana de la duración fue de 1.3 meses (rango 1 día a 8.7+ meses). La colitis condujo a discontinuación de KEYTRUDA® en 15 pacientes (0.5%). La colitis se resolvió en 41 pacientes.
Hepatitis: La mediana de tiempo hasta la aparición de hepatitis fue de 1.3 meses (rango 8 días a 21.4 meses). La mediana de la duración fue de 1.8 meses (rango 8 días a 20.9+ meses). La hepatitis condujo a la discontinuación de KEYTRUDA® en 6 pacientes (0.2%). La hepatitis se resolvió en 15 pacientes.
Nefritis: La mediana del tiempo hasta la aparición de la nefritis fue de 5.1 meses (rango 12 días a 12.8 meses). La mediana de duración fue de 3.3 meses (rango 12 días a 8.9+ meses). La nefritis condujo a la discontinuación de KEYTRUDA® en 3 pacientes (0.1%). La nefritis se resolvió en 5 pacientes.
Otros eventos adversos:
Melanoma: La Tabla 2 resume los eventos adversos que ocurrieron en al menos el 10% de los pacientes con melanoma tratado con KEYTRUDA® en KEYNOTE-006. Los eventos adversos más comunes (reportados en al menos el 15% de los pacientes) fueron artralgias y tos.
|
Tabla 2: Eventos Adversos que Ocurrieron en ≥ 10% de los Pacientes Tratados con KEYTRUDA® y con Mayor Incidencia que en el Brazo tratado con Ipilimumab (Diferencia Entre Brazo de ≥5% [Todos los Grados] o ≥2% [Grado 3]) (KEYNOTE-006) |
||||
|
KEYTRUDA® 10 mg/kg cada 2 o 3 semanas n=555 |
Ipilimumab 3 mg/kg cada 3 semanas n=256 |
|||
|
Eventos Adversos |
Todos los Grados (%) |
Grado 3* (%) |
Todos los Grados (%) |
Grado 3* (%) |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
|
Artralgia |
18 |
0 |
10 |
1 |
|
Dolor de espalda |
12 |
1 |
7 |
1 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Tos |
17 |
0 |
7 |
0 |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Vitiligo |
11 |
0 |
2 |
0 |
|
* De estos eventos adversos ≥10%, ninguno fue reportado como Grado 4. |
||||
La Tabla 3 resume los eventos adversos que ocurrieron en al menos el 10% de los pacientes con melanoma tratados con KEYTRUDA® a la dosis recomendada en KEYNOTE-002. El evento adverso más común (reportado en al menos el 20% de los pacientes) fue prurito.
|
Tabla 3: Eventos Adversos que Ocurren en ≥ 10% de los Pacientes con Melanoma Tratados con KEYTRUDA® y a una Incidencia Mayor que en el Brazo con Quimioterapia (Diferencia Entre Brazo de ≥5% [Todos los Grados] o ≥2% [Grados 3-4] ) (KEYNOTE-002) |
||||
|
KEYTRUDA® 2 mg/kg cada 3 semanas n=178 |
Quimioterapia n=171 |
|||
|
Eventos Adversos |
Todos los Grados (%) |
Grado 3-4* (%) |
Todos los Grados (%) |
Grado 3-4* (%) |
|
Trastornos gastrointestinales |
||||
|
Dolor Abdominal |
13 |
2 |
8 |
1 |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Prurito |
25 |
0 |
8 |
0 |
|
Sarpullido |
13 |
0 |
8 |
0 |
|
Trastornos de la nutrición y del metabolismo |
||||
|
Hyponatremia |
11 |
3 |
5 |
1 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
|
Artralgia |
15 |
1 |
10 |
1 |
|
* De estos eventos adversos ≥10%, ninguno fue reportado como Grado 4 en pacientes que recibieron KEYTRUDA® a dosis de 2 mg/kg. La hiponatremia fue reportada como Grado 4 en un paciente que recibió quimioterapia. |
||||
En general, el perfil de seguridad fue similar con todas las dosis y tambien fue similar entre los pacientes naïve a ipilimumab y los pacientes sin terapia previa con ipilimumab.
Carcinoma de pulmón de células no pequeñas: La Tabla 4 resume los eventos adversos que ocurrieron en al menos el 10% de los pacientes previamente tratados con NSCLC que recibieron KEYTRUDA® en KEYNOTE-010. El evento adverso más común (reportado en al menos el 15% de los pacientes) fue tos. Los eventos adversos que ocurrieron en pacientes con NSCLC no tratado previamente que recibieron KEYTRUDA® en KEYNOTE-024 fueron generalmente similares a aquellos que ocurrieron en los pacientes en KEYNOTE-010.
|
Tabla 4: Eventos Adversos que Ocurren en ≥ 10% de los Pacientes con NSCLC Tratados con KEYTRUDA® y a una Incidencia Mayor que en el Brazo con Docetaxel (Diferencia Entre Brazo de ≥5% [Todos los Grados] o ≥2% [Grado 3] ) (KEYNOTE-010) |
||||
|
KEYTRUDA® 2 o 10 mg/kg cada 3 semanas n=682 |
Docetaxel 75 mg/m2 cada 3 semanas n=309 |
|||
|
Evento Adverso |
Todos los Grados (%) |
Grado 3* (%) |
Todos los Grados (%) |
Grado 3* (%) |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Tos |
19 |
1 |
14 |
0 |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Prurito |
14 |
<1 |
7 |
0 |
|
Sarpullido |
11 |
0 |
3 |
<1 |
|
* De estos eventos adversos ≥10%, ninguno fue reportado como Grado 4. |
||||
Otros carcinomas: Los eventos adversos que ocurrieron en pacientes con HNSCC y cHL fueron generalmente similares a aquellos que ocurrieron en pacientes con melanoma o NSCLC.
INTERACCIONES MEDICAMENTOSAS Y OTRAS FORMAS DE INTERACCIÓN:
No se ha realizado ningún estudio farmacocinético formal sobre la interacción farmacológica con KEYTRUDA®. Dado que pembrolizumab se elimina de la circulación a través de catabolismo, se espera que no haya interacciones metabólicas entre medicamentos.
Se debe evitar el uso de corticoides o immunosupresores antes de iniciar el tratamiento con KEYTRUDA® debido a su posible interferencia con la actividad farmacodinámica y la eficacia de KEYTRUDA®. Sin embargo, los corticoides sistémicos y otros inmunosupresores pueden utilizarse después de iniciar KEYTRUDA® para el tratamiento de reacciones adversas mediadas inmunológicamente. [Ver Advertencias y precauciones].
ESTUDIOS CLÍNICOS:
Eficacia y seguridad clínica:
Melanoma:
KEYNOTE-006: Ensayo controlado en pacientes naïve al tratamiento con ipilimumab: La seguridad y eficacia de KEYTRUDA® fueron investigadas en KEYNOTE-006, un estudio multicéntrico, controlado, de Fase III, para el tratamiento de melanoma irresecable o metastásico en pacientes que eran naïve a ipilimumab y que no recibieron o recibieron una terapia previa sistémica. Los pacientes fueron aleatorizados (1:1:1) para recibir KEYTRUDA® en dosis de 10 mg/kg cada 2 semanas (n=279) o cada 3 semanas (n=277) o ipilimumab (n=278). La aleatorización fue estratificada por línea de terapia, estado funcional ECOG y estado de expresión de PD-L1. El estudio excluyó pacientes con enfermedad autoinmune o aquellos que recibieron inmunosupresión; hipersensibilidad severa previa a otros anticuerpos monoclonales e infección por VIH, hepatitis B o hepatitis C. Los pacientes con melanoma con mutación BRAF V600E no fueron obligados a recibir terapia previa con inhibidores BRAF.
Los pacientes fueron tratados con KEYTRUDA® hasta la progresión de la enfermedad o presencia de toxicidad inaceptable. A los pacientes clínicamente estables, con evidencia inicial de progresión de la enfermedad se les permitió permanecer en tratamiento hasta que se confirmó la progresión de la enfermedad. La evaluación del estado tumoral fue realizada a las 12 semanas, luego cada 6 semanas hasta la semana 48, seguido por cada 12 semanas de ahí en adelante.
De los 834 pacientes en KEYNOTE-006, 60% fueron hombres, 44% ≥65 años (la mediana de edad fue 62 años [rango 18-89]) y 98% fueron de raza blanca. El 66% no tenía terapia sistémica previa y por lo tanto recibieron la terapia de estudio como tratamiento de primera línea, mientras que el 34% tenían una terapia previa y por lo tanto recibieron la terapia de estudio como tratamiento de segunda línea. El 31% tenía un estado funcional ECOG de 1 y 69% un estado funcional ECOG de 0. El 80% de los pacientes eran PD-L1 positivos (expresión de membrana PD -L1 en ≥1% de células dentro de los nidos tumorales evaluados prospectivamente mediante un ensayo de investigación de inmunohistoquímica con el anticuerpo anti PD L1 22C3) y un 18% fueron PD-L1 negativos. El 65% de los pacientes tenían estadio M1c, el 32% tenía el LDH elevado y el 9% tenía metástasis cerebral. Las mutaciones BRAF se reportaron en 302 pacientes (36%). Entre los pacientes con tumores con mutación BRAF, 139 (46%) fueron tratados previamente con un inhibidor BRAF. Las características basales estuvieron bien equilibradas entre los brazos de tratamiento.
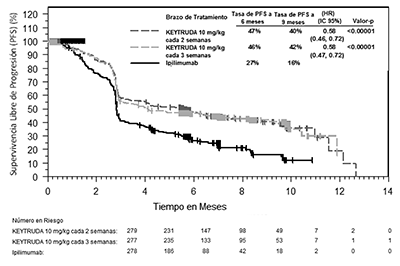
Los criterios de valoración de eficacia primaria fueron la supervivencia global (OS, por sus siglas en inglés) y la supervivencia libre de progresión (PFS, por sus siglas en inglés; según la evaluación por la revisión Integrated Radiology and Oncology Assessment [IRO] utilizando los Criterios de Evaluación de Respuesta en los Tumores Sólidos [RECIST 1.1, por sus siglas en inglés]). Los criterios de valoración de eficacia secundaria fueron la tasa de respuesta global (TRG) y la duración de la respuesta. La Tabla 5 resume las principales medidas de eficacia y las curvas de Kaplan-Meier para la supervivencia global y la supervivencia libre de progresión se muestran en las Figuras 1 y 2.
|
Tabla 5: Respuesta a KEYTRUDA® 10 mg/kg cada 2 o 3 semanas en pacientes con melanoma avanzado naïve a KEYTRUDA®, en KEYNOTE-006 |
|||
|
Criterios de Valoración |
KEYTRUDA® 10 mg/kg cada 3 semanas n=277 |
KEYTRUDA® 10 mg/kg cada 2 semanas n=279 |
Ipilimumab n=278 |
|
OS (supervivencia global) |
|||
|
Número (%) de pacientes con evento |
92 (33%) |
85 (30%) |
112 (40%) |
|
Cociente de riesgo † (IC 95%) |
0.69 (0.52, 0.90) |
0.63 (0.47, 0.83) |
- - - |
|
Valor-p ‡ |
0.00358 |
0.00052 |
- - - |
|
Mediana en meses (IC del 95%) |
No alcanzado (NA, NA) |
No alcanzado (NA, NA) |
No alcanzado (13, NA) |
|
PFS mediante IRO* |
|||
|
Número (%) de pacientes con evento |
157 (57%) |
157 (56%) |
188 (68%) |
|
Cociente de riesgo † (IC del 95%) |
0.58 (0.47, 0.72) |
0.58 (0.46, 0.72) |
- - - |
|
Valor-p ‡ |
<0.00001 |
<0.00001 |
- - - |
|
Mediana en meses (IC del 95%) |
4.1 (2.9, 6.9) |
5.5 (3.4, 6.9) |
2.8 (2.8, 2.9) |
|
Mejor respuesta general mediante IRO* |
|||
|
TRG % (IC del 95%) |
33% (27, 39) |
34% (28, 40) |
12% (8, 16) |
|
Respuesta completa % |
6% |
5% |
1% |
|
Respuesta parcial % |
27% |
29% |
10% |
|
Duración de la respuesta § mediante IRO* |
|||
|
Mediana en meses (rango) |
No alcanzado (1.4+, 8.1+) |
8.3 (1.4+, 8.3) |
No alcanzado (1.1+, 7.9+) |
|
% en curso |
97% |
89% |
88% |
|
* IRO = Revisión radiológica independiente más oncológica utilizando RECIST 1.1 † Cociente de riesgo (KEYTRUDA® en comparación a ipilimumab) basado en el modelo de riesgo proporcional de Cox estratificado. ‡ Basado en la prueba Log Rank estratificada. § Basado en los pacientes con una respuesta general mejor como se confirma por la respuesta completa o parcial. |
|||
Figura 1: Curva de Kaplan-Meier para la supervivencia global por brazo de tratamiento, en KEYNOTE-006 (población con intención de tratamiento).
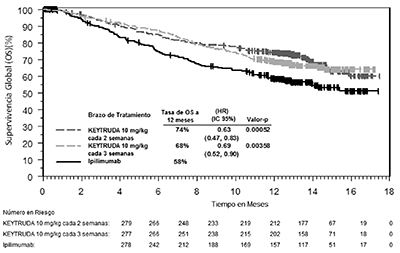
Figura 2: Curva de Kaplan-Meier para la supervivencia libre de progresión (con base en IRO), por brazo de tratamiento, en KEYNOTE-006 (población con intención de tratamiento).
Análisis de subpoblación por estado de mutación BRAF: Se realizó un análisis de subgrupo de KEYNOTE-006 en los pacientes que tenían mutación BRAF tipo silvestre, mutación BRAF sin tratamiento BRAF previo, y mutación BRAF con tratamiento BRAF previo. Los cocientes de riesgo (HRs) de PFS (KEYTRUDA® agrupado [10 mg/kg cada 2 o 3 semanas] vs. ipilimumab) fueron 0.57 (IC del 95%: 0.45, 0.73) para el BRAF tipo silvestre, 0.50 (IC del 95%: 0.32, 0,77) para la mutación BRAF sin tratamiento previo de BRAF y 0.73 (IC del 95%: 0.48,
1.11) para la mutación BRAF con tratamiento previo de BRAF. Los HRs de la OS para KEYTRUDA® agrupado vs. ipilimumab fueron 0.61 (IC del 95%: 0.46, 0,82) para BRAF tipo silvestre, 0.69 (IC del 95%: 0.33, 1.45) para la mutación BRAF sin tratamiento previo de BRAF), y 0.75 (IC del 95%: 0.45, 1.26) para la mutación BRAF con tratamiento previo de BRAF. La TRG para KEYTRUDA® agrupado vs. ipilimumab fue del 34% vs. 13% para BRAF tipo silvestre, 41% vs. 13% para la mutación BRAF sin tratamiento previo de BRAF y 21% vs. 6% para la mutación BRAF con tratamiento previo de BRAF.
Análisis de subpoblación por estado PD-L1: Se realizó un análisis de subgrupo de KEYNOTE 006 en los pacientes PD L1 positivos vs. los pacientes PD L1 negativos. Los HRs de la PFS (KEYTRUDA® agrupado [10 mg/kg cada 2 o 3 semanas] vs. ipilimumab) fueron 0.53 (IC del 95%: 0.43, 0.65) para pacientes PD L1 positivos y 0.73 (IC del 95%: 0.47, 1.11) para pacientes PD L1 negativos. Los HRs de la OS para el grupo KEYTRUDA® agrupado vs. ipilimumab fueron 0.56 (IC del 95%: 0.43, 0.73) para los pacientes PD L1 positivos y 0.95 (IC del 95%: 0.56, 1.62) para los pacientes PD L1 negativos.
KEYNOTE-002:
Ensayo controlado en pacientes con melanoma previamente tratados con ipilimumab: La seguridad y eficacia de KEYTRUDA® fueron investigadas en KEYNOTE-002, un estudio controlado, multicéntrico, para el tratamiento del melanoma irresecable o metastásico en pacientes previamente tratados con ipilimumab y en casos positivos para la mutación BRAF V600, con un inhibidor de BRAF o MEK. Los pacientes fueron aleatorizados (1:1:1) para recibir KEYTRUDA® a una dosis de 2 mg/kg (n=180) o 10 mg/kg (n=181) cada 3 semanas o quimioterapia (n=179; incluyendo dacarbazina, temozolomida, carboplatino, paclitaxel o carboplatino+paclitaxel). El estudio excluyó pacientes con enfermedad autoinmune o que recibieron inmunosupresión; un antecedente de reacciones adversas mediadas inmunológicamente severas o potencialmente mortales dell tratamiento con ipilimumab, definidas como cualquier toxicidad Grado 4 o Grado 3 que requirió tratamiento con corticoides (mayor a 10 mg/día de prednisona o su dosis equivalente) durante más de 12 semanas; hipersensibilidad severa previa para otros anticuerpos monoclonales; un antecedente de neumonitis o de enfermedad pulmonar intersticial; Infección por VIH, hepatitis B o hepatitis C.
Los pacientes fueron tratados con KEYTRUDA® hasta la progresión de la enfermedad o presencia de toxicidad inaceptable. A los pacientes clínicamente estables, con evidencia inicial de progresión de la enfermedad se les permitió permanecer en el tratamiento hasta que se confirmó la progresión de la enfermedad. La evaluación del estado tumoral fue realizada a las 12 semanas, luego cada 6 semanas hasta la Semana 48, seguido por cada 12 semanas de ahí en adelante. Los pacientes en quimioterapia que experimentaron progresión de la enfermedad verificada independientemente, después de la primera evaluación programada de la enfermedad, fueron capaces de cruzar y recibir 2 mg/kg o 10 mg/kg de KEYTRUDA® cada 3 semanas en un diseño doble ciego.
De los 540 pacientes en KEYNOTE-002, 61% fueron hombres, 43% fueron ≥65 años (la mediana de edad fue de 62 años [rango 15-89]) y el 98% fueron de raza blanca blanca. El 82% de los pacientes tenían estadio M1c, el 73% tenía al menos y el 32% tenía tres o más terapias sistémicas previas para el melanoma avanzado. El 45% tenía una ECOG PS de 1, el 40% LDH elevada y el 23% tenía un tumor con mutación BRAF. Las características basales fueron bien equilibradas entre los brazos de tratamiento.
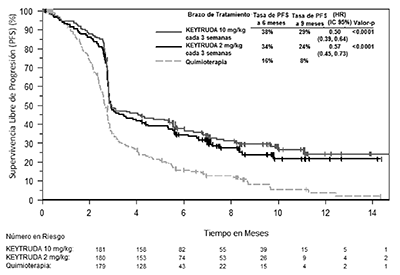
Las criterios de valoración de eficacia primaria fueron la PFS (según la evaluación por la IRO usando RECIST 1.1) y la OS. Los criterios de valoración de eficacia secundaria fueron PFS (según la evaluación por el investigador utilizando RECIST 1.1), TRG y duración de la respuesta. La Tabla 6 resume las medidas clave de eficacia en pacientes previamente tratados con ipilimumab y la curva de Kaplan-Meier para la PFS se muestra en la Figura 3. Los datos de la OS no estaban disponibles en el momento de los análisis de la PFS. No hubo ninguna diferencia estadísticamente significativa entre KEYTRUDA® y la quimioterapia en el análisis preliminar de la OS que no fue ajustado para los efectos de confusión potenciales por el cambio. De los pacientes aleatorizados en el brazo con quimioterapia, el 48% cambió y recibió posteriormente tratamiento con KEYTRUDA®.
|
Tabla 6: Respuesta a KEYTRUDA® 2 mg/kg o 10 mg/kg cada 3 semanas en pacientes con melanoma irresecable o metastásico en KEYNOTE-002 |
|||
|
Criterio de Valoración |
KEYTRUDA® 2 mg/kg cada 3 semanas n=180 |
KEYTRUDA® 10 mg/kg cada 3 semanas n=181 |
Quimioterapia n=179 |
|
PFS por medio de IRO* |
|||
|
Número (%) de pacientes con evento |
129 (72%) |
126 (70%) |
155 (87%) |
|
Cociente de riesgo† (IC del 95%) |
0.57 (0.45, 0.73) |
0.50 (0.39, 0.64) |
- - - |
|
Valor-p ‡ |
<0.0001 |
<0.0001 |
- - - |
|
Mediana en meses (IC del 95%) |
2.9 (2.8, 3.8) |
2.9 (2.8, 4.7) |
2.7 (2.5, 2.8) |
|
Promedio en meses (IC del 95%)§ |
5.4 (4.7, 6.0) |
5.8 (5.1, 6.4) |
3.6 (3.2, 4.1) |
|
PFS por INV¶ |
|||
|
Número (%) de pacientes con evento |
122 (68%) |
112 (62%) |
157 (88%) |
|
Cociente de riesgo† (IC del 95%) |
0.49 (0.38, 0.62) |
0.41 (0.32, 0.52) |
- - - |
|
Valor-p ‡ |
<0.0001 |
<0.0001 |
- - - |
|
Mediana en meses (IC del 95%) |
3.7 (2.9, 5.4) |
5.4 (3.8, 6.8) |
2.6 (2.4, 2.8) |
|
Promedio en meses (IC del 95%) § |
5.8 (5.2, 6.4) |
6.5 (5.8, 7.1) |
3.7 (3.2, 4.1) |
|
Mejor respuesta general por IRO* |
|||
|
TRG % (IC del 95%) |
21% (15, 28) |
25% (19, 32) |
4% (2, 9) |
|
Respuesta completa % |
2% |
3% |
0% |
|
Respuesta parcial % |
19% |
23% |
4% |
|
Duración de la respuesta # por IRO* |
|||
|
Mediana en meses (rango) |
No alcanzado (1.4+, 11.5+) |
No alcanzado (1.2+, 11.1+) |
8.5 (1.6+, 9.5) |
|
% en curso |
87% |
80% |
63% |
|
* IRO = Revisión radiología independiente más oncología utilizando RECIST 1.1. † Cociente de riesgo (KEYTRUDA® en comparación con ipilimumab) basado en el modelo de riesgo proporcional de Cox estratificado. ‡ Basado en la prueba Log Rank estratificada. § Tiempo de sobrevida libre de progresión promedio restringido, con base en el seguimiento de 12 meses. ¶ INV = Evaluación del investigador utilizando RECIST 1.1. # Basado en los pacientes con una respuesta general mejor como se confirma por la respuesta completa o parcial. |
|||
Figura 3: Curva de Kaplan-Meier para la supervivencia libre de progresión (basada en IRO), por brazo de tratamiento, en KEYNOTE-002 (población con intención de tratamiento).
KEYNOTE-001: Estudio de etiqueta abierta en pacientes con melanoma: También se investigó la seguridad y eficacia de KEYTRUDA® en un estudio no controlado, de etiqueta abierta, para el tratamiento del melanoma irresecable o metastásico. Se evaluó la eficacia en 276 pacientes de dos cohortes de KEYNOTE-001, uno que incluyó a pacientes que previamente fueron trataron con ipilimumab (y si eran positivos para la mutación BRAF V600, un inhibidor de BRAF o MEK) y otro cohorte que incluyó pacientes naïve al tratamiento con ipilimumab. Los pacientes fueron aleatorizados para recibir KEYTRUDA® a una dosis de 2 mg/kg cada 3 semanas o 10 mg/kg cada 3 semanas. Los pacientes fueron tratados con KEYTRUDA® hasta la progresión de la enfermedad o presencia de toxicidad inaceptable. A los pacientes clínicamente estables, con evidencia inicial de progresión de la enfermedad se les permitió permanecer en tratamiento hasta que se confirmó la progresión de la enfermedad. Los criterios de exclusión fueron similares a los de KEYNOTE 002.
De los 89 pacientes que recibieron 2 mg/kg de KEYTRUDA® y fueron previamente tratados con ipilimumab, 53% fueron hombres, 33% fueron ≥65 años de edad y la edad promedio fue de 59 años (rango 18-88). Todos, menos dos pacientes, fuero de raza blanca. El 84% de los pacientes tenía estadio M1c y 8% tenía un antecedente de metástasis cerebral. El 78% de los pacientes tenían al menos dos y el 35% tenía tres o más terapias sistémicas previas para melanoma avanzado. Las mutaciones BRAF fueron registradas en el 13% de la población del estudio.
De los 51 pacientes que recibieron 2 mg/kg de KEYTRUDA® que eran naïve al tratamiento con ipilimumab, el 63% fueron hombres, el 35% fueron ≥65 años de edad y la mediana de edad fue de 60 años (rango 35-80). Todos menos un paciente fue de raza blanca. El 63% de los pacientes tenían
estadio M1c y el 2% tenía un antecedente de metástasis cerebral. El 45% no tenía terapias previas para melanoma avanzado. Las mutaciones BRAF fueron registradas en el 39% de la población del estudio.
El criterio de valoración de eficacia primario fue la TRG según la evaluación de la revisión independiente utilizando las respuestas confirmadas y RECIST 1.1. Los criterios de valoración de eficacia secundaria fueron la tasa de control de la enfermedad (TCE; incluyendo respuesta completa, respuesta parcial y enfermedad estable), duración de la respuesta, PFS y OS. La respuesta tumoral se evaluó en intervalos de 12 semanas. La Tabla 7 resume las medidas clave de eficacia en los pacientes previamente tratados o naïve al tratamiento con ipilimumab, que recibieron KEYTRUDA® a la dosis recomendada.
|
Tabla 7: Respuesta a KEYTRUDA® 2 mg/kg cada 3 semanas en pacientes con melanoma irresecable o metastásico en KEYNOTE-001 |
||
|
Criterio de Valoración |
KEYTRUDA® 2 mg/kg cada 3 semanas en pacientes previamente tratados con ipilimumab n=89 |
KEYTRUDA® 2 mg/kg cada 3 semanas en pacientes naïve al tratamiento con ipilimumab n=51 |
|
Mejor Respuesta General* por IRO† |
||
|
TRG %, (IC del 95%) |
25% (16, 35) |
33% (21, 48) |
|
Tasa de Control de la Enfermedad %‡ |
49% |
49% |
|
Respuesta Completa |
3% |
10% |
|
Respuesta Parcial |
21% |
24% |
|
Enfermedad Estable |
25% |
16% |
|
Duración de la Respuesta§ |
||
|
Mediana en meses (rango) |
No alcanzado (2.8+, 14.3+) |
No alcanzado (1.6+, 13.8+) |
|
% en curso |
86%¶ |
82%# |
|
PFS |
||
|
Mediana en meses (IC del 95%) |
4.9 (2.8, 8.3) |
5.5 (2.8, 14.0) |
|
Tasa de PFS a los 6 meses |
43% |
50% |
|
OS |
||
|
Mediana en meses (IC del 95%) |
No alcanzado (11, no disponible) |
No alcanzado (14, no disponible) |
|
Tasa de OS a los 12 meses |
60% |
72% |
|
* Incluye pacientes sin enfermedad medible en al inicio del estudio mediante radiología independiente. † IRO = Revisión radiológica independiente más oncología utilizando RECIST 1.1. ‡ Basado en la mejor respuesta de la enfermedad estable o mejor. § Basado en pacientes con una respuesta confirmada por revisión independiente, a partir de la fecha en la cual la respuesta se registró por primera vez; n = 22 para pacientes previamente tratados con Ipilimumab; n = 17 para pacientes naïve al tratamiento con ipilimumab. ¶ Los respondedores fueron seguidos por al menos 12 meses después del inicio de la terapia. # Los respondedores fueron seguidos por al menos 15 meses después del inicio de la terapia. |
||
Los resultados para los pacientes previamente tratados con ipilimumab (n=84) y naïve al tratamiento con ipilimumab (n=52) que recibieron 10 mg/kg de KEYTRUDA® cada 3 semanas fueron similares a los observados en los pacientes que recibieron 2 mg/kg de KEYTRUDA® cada 3 semanas.
Carcinoma de pulmón de células no pequeñas:
KEYNOTE-024:
Ensayo controlado de pacientes con NSCLC no tratados previamente: La eficacia de KEYTRUDA® se investigó en KEYNOTE-024, un ensayo multicéntrico, aleatorizado y controlado. Los criterios de elegibilidad clave fueron NSCLC metastásico, TPS de expresión de PD-L1 de 50% o más mediante un ensayo de inmunohistoquímica usando el kit PD-L1 de IHC 22C3 pharmDx y ningún tratamiento sistémico previo para NSCLC metastásico. Pacientes con aberraciones tumorales genómicas de EGFR o ALK; enfermedad autoinmune que requirió terapia sistémica dentro de los 2 años de tratamiento; una condición médica que requirió inmunosupresión; o quienes habían recibido más de 30 G y de radiación torácica dentro de las 26 semanas anteriores no eran elegibles. Los pacientes fueron asignados al azar (1: 1) para recibir KEYTRUDA® 200 mg cada 3 semanas (n = 154) o la elección del investigador de quimioterapia con platino (n = 151, incluyendo pemetrexed + carboplatino, pemetrexed + cisplatino, gemcitabina + cisplatino, gemcitabina + o paclitaxel + carboplatino. Los pacientes con carcinoma no escamoso podrían recibir mantenimiento con pemetrexed). Los pacientes fueron tratados con KEYTRUDA® hasta toxicidad inaceptable o progresión de la enfermedad.
El tratamiento podría continuar más allá de la progresión de la enfermedad si el paciente estuviera clínicamente estable y se considerara que obtendría beneficios clínicos por parte del investigador. Los pacientes sin progresión de la enfermedad podrían ser tratados por hasta 24 meses. El tratamiento con KEYTRUDA® podría reiniciarse por progresión de la enfermedad posterior y administrarse hasta un año más. La evaluación del estado del tumor se realizó cada 9 semanas. Los pacientes en quimioterapia que experimentaron progresión independientemente comprobada de la enfermedad fueron capaces de cambiar y recibir KEYTRUDA®.
Entre los 305 pacientes en KEYNOTE-024, las características basales fueron: edad promedio 65 años (54% de 65 años o más); 61% varones; 82% blancos y 15% asiáticos; y 35% y 65% con un estado de desempeño ECOG 0 y 1, respectivamente. Las características de la enfermedad fueron escamosas (18%) y no escamosas (82%); M1 (99%); y metástasis cerebrales (9%).
El criterio de valoración de eficacia primaria fue PFS evaluada por la revisión central independiente ciega (BICR, por sus siglas en inglés) utilizando RECIST 1.1. Los criterios de valoración de eficacia secundaria fueron OS y ORR (evaluado por BICR usando RECIST 1.1). La Tabla 8 resume las medidas claves de eficacia para toda la población ITT.
|
Tabla 8: Resultados de Eficacia en KEYNOTE-024 |
||
|
Endpoint |
KEYTRUDA® 200 mg cada 3 semanas n=154 |
Quimioterapia n=151 |
|
PFS* |
||
|
Número (%) de pacientes con evento |
73 (47%) |
116 (77%) |
|
Hazard ratio† (95% CI) |
0.50 (0.37, 0.68) |
|
|
Valor-p ‡ |
<0.001 |
|
|
Mediana en meses (95% CI) |
10.3 (6.7, ND) |
6.0 (4.2, 6.2) |
|
OS |
||
|
Número (%) de pacientes con evento t |
44 (29%) |
64 (42%) |
|
Hazard ratio† (95% CI) |
0.60 (0.41, 0.89) |
|
|
Valor-p ‡ |
0.005 |
|
|
Mediana en meses (95% CI) |
No alcanzado (ND, ND) |
No alcanzado (9.4, ND) |
|
Tasa de respuesta objetiva* |
||
|
ORR % (95% CI) |
45% (37, 53) |
28% (21, 36) |
|
Respuesta Completa % |
4% |
1% |
|
Respuesta Parcial % |
41% |
27% |
|
Duración de la respuesta§ |
||
|
Mediana en meses (rango) |
No alcanzado (1.9+, 14.5+) |
6.3 (2.1+, 12.6+) |
|
% con duración ≥ 6 meses |
88%¶ |
59%# |
|
* Evaluado por BICR usando RECIST 1.1. † Hazard ratio (KEYTRUDA® comparado con quimioterapia) basado en el Modelo de riesgo proporcional Cox estratificado. ‡ Basado en una prueba de rango logarítmica estratificada. § Basado en pacientes con una mejor respuesta global como respuesta completa o parcial confirmada. ¶ Basado en estimados Kaplan-Meier; incluye 43 pacientes con respuestas de 6 meses o más. # Basado en estimados Kaplan-Meier; incluye 16 pacientes con respuestas de 6 meses o más. ND = no disponible. |
||
Figura 4: Curva de Kaplan-Meier para la supervivencia libre de progresión por brazo de tratamiento en KEYNOTE-024 (población con intención de tratamiento).
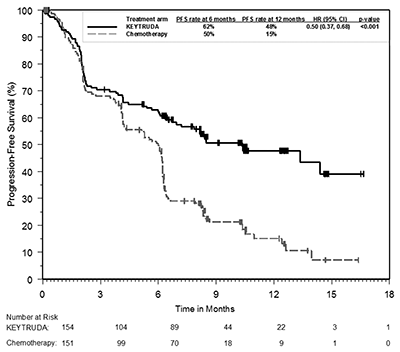
Figura 5: Curva de Kaplan-Meier para la supervivencia general por brazo de tratamiento en KEYNOTE-024 (Población con intención de tratamiento).
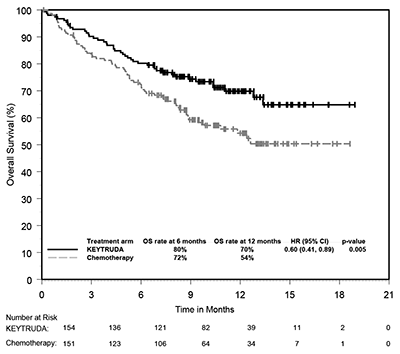
El beneficio mejorado evaluado por PFS, OS, ORR y duración de la respuesta para KEYTRUDA® en comparación con la quimioterapia en la población estudiada se asoció con mejoras en la calidad de vida relacionada con la salud (CVRS). El cambio desde la línea de base hasta la semana 15 mostró una mejora significativa en el Cuestionario de Calidad de Vida de la Organización Europea de Investigación y Tratamiento del Cáncer (EORTC QLQ) C30 estado de salud global / puntuación de QoL para los pacientes que recibieron KEYTRUDA® en comparación con la quimioterapia (diferencia en LS media = 7.82, 95% IC: 2,85, 12,79, dos lados p = 0,002). El tiempo hasta el deterioro en el EORTC QLQ-LC13 punto final compuesto de tos, disnea y dolor torácico fue prolongado para los pacientes que recibieron KEYTRUDA® en comparación con la quimioterapia (HR = 0,66, 95% IC: 0,44,
0,97, dos lados p= 0,029) donde el deterioro se define como una disminución confirmada de 10 puntos o mas desde la línea de base en cualquiera de estos tres síntomas.
KEYNOTE-010:
Ensayo controlado de pacientes NSCLC previamente tratados con quimioterapia: Se investigó la eficacia de KEYTRUDA® en KEYNOTE-010, un ensayo multicéntrico, aleatorizado y controlado. Los criterios clave de elegibilidad fueron NSCLC avanzados que habían progresado después de la quimioterapia con platino, y si fuere apropiado, terapia dirigida para las mutaciones ALK o EGFR y PPT con expresión de PD-L1 de 1% o más mediante una versión de ensayo clínico del kit PD-L1 IHC 22C3 pharmDxTM. Fueron inelegibles los pacientes con enfermedad autoinmune; una condición médica que requiere inmunosupresión; o que hayan recibido más de 30 Gy de radiación torácica dentro de las últimas 26 semanas. Los pacientes fueron aleatorizados (1:1:1) para recibir 2 mg/kg (n=344) o 10 mg/kg (n=346) de KEYTRUDA® cada 3 semanas o 75 mg/m2 de docetaxel cada 3 semanas (n=343). Los pacientes fueron tratados con KEYTRUDA® hasta la progresión de la enfermedad o presencia de toxicidad inaceptable. Se realizó evaluación del estado del tumor cada 9 semanas.
Entre los 1033 pacientes en KEYNOTE-010, las características iniciales fueron: edad media 63 años (42% de 65 años o mayores); 61% hombres; 72% Blancos y 21% Asiáticos; y 34% y 66% con un estado funcional ECGO 0 y 1, respectivamente. Las características de la enfermedad fueron escamosas (21%) y no escamosas (70%); M1 (91%); metástasis cerebral (15%); y la incidencia de aberraciones genómicas fue EGFR (8%) o ALK (1%). El tratamiento previo incluyó régimen doble de platino (100%); los pacientes recibieron una (69%), o dos o más (29%) de terapias previas.
Los criterios de valoración de eficacia primaria fueron la OS y la PFS según lo evaluado por un comité de revisión independiente usando RECIST 1.1. Los criterios de valoración de eficacia secundaria fueron TRG y la duración de la respuesta. La Tabla 9 resume las medidas de eficacia clave para toda la población ITT (PPT ≥1) y para el subgrupo de pacientes con PPT ≥50%. Las curvas de Kaplan-Meier para la OS (PPT ≥1% y PPT ≥50%) se muestran en las Figuras 6 y 7).
|
Tabla 9: Respuesta a KEYTRUDA® 2 o 10 mg/kg cada 3 Semanas en Pacientes Previamente tratados con NSCLC en KEYNOTE-010 |
|||
|
Criterio de Valoración |
KEYTRUDA® 2 mg/kg cada 3 semanas |
KEYTRUDA® 10 mg/kg cada 3 semanas |
Docetaxel 75 mg/m2 cada 3 semanas |
|
PPT ≥1% |
|||
|
Número de pacientes |
344 |
346 |
343 |
|
OS |
|||
|
Número (%) de pacientes con evento |
172 (50%) |
156 (45%) |
193 (56%) |
|
Cociente de riesgo* (IC del 95%) |
0.71 (0.58, 0.88) |
0.61 (0.49, 0.75) |
- - - |
|
Valor-p† |
<0.001 |
<0.001 |
- - - |
|
Mediana en meses (IC del 95%) |
10.4 (9.4, 11.9) |
12.7 (10.0, 17.3) |
8.5 (7.5, 9.8) |
|
PFS‡ |
|||
|
Número (%) de pacientes con evento |
266 (77%) |
255 (74%) |
257 (75%) |
|
Cociente de riesgo* (IC del 95%) |
0.88 (0.73, 1.04) |
0.79 (0.66, 0.94) |
- - - |
|
Valor-p† |
0.068 |
0.005 |
- - - |
|
Mediana en meses (IC del 95%) |
3.9 (3.1, 4.1) |
4.0 (2.6, 4.3) |
4.0 (3.1, 4.2) |
|
Tasa de respuesta global‡ |
|||
|
TRG %§ (IC del 95%) |
18% (14, 23) |
18% (15, 23) |
9% (7, 13) |
|
Duración de la respuesta ‡,¶,# |
|||
|
Mediana en meses (rango) |
No alcanzado (0.7+, 20.1+) |
No alcanzado (2.1+, 17.8+) |
6.2 (1.4+, 8.8+) |
|
% en curso |
73% |
72% |
34% |
|
PPT ≥50% |
|||
|
Número de pacientes |
139 |
151 |
152 |
|
OS |
|||
|
Número (%) de pacientes con evento |
58 (42%) |
60 (40%) |
86 (57%) |
|
Cociente de riesgo* (IC del 95%) |
0.54 (0.38, 0.77) |
0.50 (0.36, 0.70) |
- - - |
|
Valor-p † |
<0.001 |
<0.001 |
- - - |
|
Mediana en meses (IC del 95%) |
14.9 (10.4, NA) |
17.3 (11.8, NA) |
8.2 (6.4, 10.7) |
|
PFS‡ |
|||
|
Número (%) de pacientes con evento |
89 (64%) |
97 (64%) |
118 (78%) |
|
Cociente de riesgo* (IC del 95%) |
0.58 (0.43, 0.77) |
0.59 (0.45, 0.78) |
- - - |
|
Valor-p† |
<0.001 |
<0.001 |
- - - |
|
Mediana en meses (IC del 95%) |
5.2 (4.0, 6.5) |
5.2 (4.1, 8.1) |
4.1 (3.6, 4.3) |
|
Tasa de respuesta global‡ |
|||
|
TRG %§ (IC del 95%) |
30% (23, 39) |
29% (22, 37) |
8% (4, 13) |
|
Duración de la respuesta ‡,¶,Þ |
|||
|
Mediana en meses (rango) |
No alcanzado (0.7+, 16.8+) |
No alcanzado (2.1+, 17.8+) |
8.1 (2.1+, 8.8+) |
|
% en curso |
76% |
75% |
33% |
|
* Cociente de riesgo (KEYTRUDA® en comparación con docetaxel) basado en el modelo de riesgo proporcional de Cox estratificado. † Basado en la prueba Log rank estratificada. ‡ Evaluado por BICR usando RECIST 1.1. § Todas las respuestas fueron respuestas parciales. ¶ Basado en los pacientes con una respuesta general mejor como se confirma por la respuesta completa o parcial. # Incluye 30, 31, y 2 pacientes con respuestas en curso de 6 meses o más en los grupos KEYTRUDA® 2 mg/kg, KEYTRUDA® 10 mg/kg, y docetaxel, respectivamente. Þ Incluye 22, 24, y 1 paciente con respuestas en curso de 6 meses o más en los grupos KEYTRUDA® 2 mg/kg, KEYTRUDA® 10 mg/kg, y docetaxel, respectivamente. |
|||
Figura 6: Curva de Kaplan-Meier para la Supervivencia Global por Brazo de Tratamiento, en KEYNOTE-010 (PPT ≥1%, Población con Intención de Tratamiento).
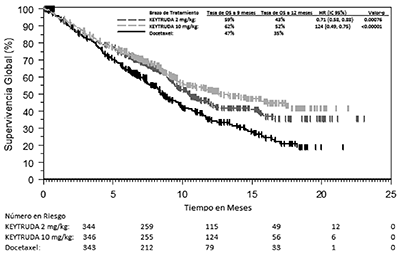
Figura 7: Curva de Kaplan-Meier para la Supervivencia Global por Brazo de Tratamiento, en KEYNOTE-010 (PPT ≥50%, Población con Intención de Tratamiento).
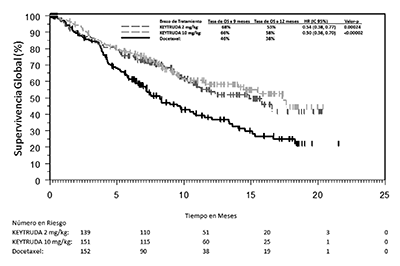
Los resultados de eficacia fueron similares para los brazos de 2 mg/kg y 10 mg/kg de KEYTRUDA®. Los resultados de eficacia para la OS fueron consistentes independientemente de la edad de la muestra del tumor (nuevos vs. archivos).
KEYNOTE-001:
Estudio de etiqueta abierta en pacientes con NSCLC tratados previamente con quimioterapia: También se investigó la eficacia de KEYTRUDA® en un cohorte comparativo de dosis de KEYNOTE-001 aleatorizado, de etiqueta abierta, multicéntrico. Los pacientes tenían NSCLC avanzado que era PD-L1 positivo, con progresión de la enfermedad después del tratamiento con quimioterapia con platino. Los pacientes con aberraciones genómicas tumorales EGFR o ALK tuvieron progresión de la enfermedad durante la terapia aprobada para estas aberraciones antes de recibir KEYTRUDA®. El ensayo excluyó pacientes con enfermedad autoinmune; una condición médica que requiere inmunosupresión; o que han recibido más de 30 G y de radiación torácica dentro de las últimas 26 semanas. Los pacientes fueron aleatorizados para recibir 10 mg/kg de KEYTRUDA® cada 2 (n=69) o cada 3 (n=87) semanas hasta progresión de la enfermedad o presencia de toxicidad inaceptable. La evaluación del estado tumoral fue realizada cada 9 semanas. Los criterios de valoración de eficacia principales fueron la tasa de respuesta global (TRG) (de acuerdo con RECIST 1.1, según la evaluación de la revisión central independiente cegada) y duración de la respuesta.
La prevalencia de pacientes con una PPT de expresión de PD-L1 mayor o igual al 50% entre los pacientes examinados con NSCLC, de acuerdo con la comprobación retrospectiva con el kit de diagnóstico PD-L1 IHC 22C3 pharmDx™ fue del 26%. Entre los pacientes aleatorizados con muestras tumorales evaluables para la expresión de PD-L1, 61 tenían PPT mayor o igual al 50%. Las características iniciales para esta población incluyeron: mediana de edad 60 años (34 de 65 años o mayores); 61% hombres; 79% Blancos; y el 34% y 64% con un estado funcional ECOG 0 y 1, respectivamente.
Las características de la enfermedad fueron escamosas y no escamosas (21% y 75%, respectivamente); M1 (98%); metástasis cerebral (11%); y una terapia previa (25%), dos (31%), o tres o más terapias previas (44%). El estado de mutación entre pacientes fue EGFR (10%), ALK (0%), o Kras (16%).
Los resultados de eficacia para los pacientes de NSCLC tratados con 10 mg/kg cada 2 o 3 semanas en KEYNOTE-001 se resumen en la Tabla 10.
|
Tabla 10: Respuesta a KEYTRUDA® 10 mg/kg cada 2 o 3 Semanas en los Pacientes con NSCLC Previamente Tratados con PPT de la expresión de PD-L1 ≥50% (n=61) |
|
|
Criterio de Valoración |
|
|
Mejor respuesta general * |
|
|
TRG %, (IC del 95%) |
43% (30, 56) |
|
Respuesta completa |
2% |
|
Repuesta parcial |
41 % |
|
Duración de la Respuesta† |
|
|
Mediana en meses (rango) |
No alcanzado (2.1+, 13.4+) |
|
% en curso |
65%‡ |
|
Tiempo de respuesta† |
|
|
Mediana en meses (rango) |
2.1 (1.4, 6.2) |
|
PFS§ |
|
|
Mediana en meses (IC del 95%) |
6.3 (2.1, 10.7) |
|
Tasa de PFS a 6 meses |
53% |
|
OS§ |
|
|
Tasa de OS a 12 meses |
60% |
|
* Basado en todos los pacientes tratados (n=61), con evaluación por revisión independiente y RECIST 1.1. † Con base en los pacientes (n=26) con respuesta confirmada por revisión independiente. ‡ Incluye 17 pacientes con respuestas en curso de 6 meses o más. § Con base en todos los pacientes tratados (n=61). |
|
Resultados similares de TRG fueron observados en otro grupo de pacientes (n=25) con PPT mayor o igual al 50% de los que recibieron KEYTRUDA® en dosis de 2 mg/kg cada 3 semanas en KEYNOTE-001.
Inmunogenicidad: En estudios clínicos en pacientes tratados con pembrolizumab con dosis de 2 mg/kg cada 3 semanas o 10 mg/kg cada dos o tres semanas, 19 (1.7%) de 1087 pacientes evaluables dieron positivo para anticuerpos de tratamiento emergente contra pembrolizumab durante el tratamiento con KEYTRUDA®. No hubo pruebas de un perfil farmacocinético o de seguridad alterado con el desarrollo de anticuerpos de unión anti-pembrolizumab.
FARMACOLOGÍA CLÍNICA:
Clase terapéutica: KEYTRUDA® (pembrolizumab) es un anticuerpo monoclonal, agente antineoplásico.
Mecanismo de acción: El PD-1 es un receptor de verificación inmune que limita la actividad de los linfocitos T en los tejidos periféricos. La vía del PD-1 es un punto de control inmunológico que puede ser comprometido por las células tumorales para inhibir la vigilancia inmunológica de las células T activas. KEYTRUDA® es un anticuerpo de alta afinidad contra PD-1, que ejerce bloqueo dual de ligandos de la vía del PD-1, incluyendo PD-L1 y PD-L2 en células presentadores de antígeno o en células tumorales. Al inhibir la unión del receptor PD-1 a sus ligandos, KEYTRUDA® reactiva los linfocitos T citotóxicos específicos de tumores en el microambiente tumoral y reactiva la inmunidad antitumoral.
ADVERTENCIAS Y PRECAUCIONES:
Reacciones adversas mediadas inmunológicamente: Se han presentado reacciones adversas mediadas inmunológicamente en pacientes que recibieron KEYTRUDA®. En los ensayos clínicos, la mayoria de las reacciones adversas mediadas inmunológicamente fueron reversibles y fueron manejadas con interrupciones de KEYTRUDA®, administración de corticoides y/o con tratamiento de apoyo. Reacciones adversas inmunomediadas que afectan mas de un sistema corporal, pueden ocurrir simultáneamente.
Cuando se sospechan reacciones adversas mediadas inmunológicamente, se debe garantizar una evaluación adecuada para confirmar la etiología o excluir otras causas. Con base en la severidad de la reacción adversa, suspender KEYTRUDA® y administrar corticoides (ver a continuación). Después de mejorar a Grado 1 o menos, iniciar la disminución de corticoides y continuar la disminución gradual durante al menos 1 mes. Con base en datos limitados de los estudios clínicos en pacientes cuyas reacciones adversas relacionadas inmunológicamente no pudieron ser controladas con el uso de corticoides, puede considerarse la administración de otros inmunosupresores sistémicos. Reiniciar KEYTRUDA® si la reacción adversa permanece en Grado 1 o menos siguiendo la disminución de corticoides. Si se produce otro episodio de reacción adversa severa, suspenda KEYTRUDA® permanentemente. [Ver Dosis y administración y Reacciones adversas].
Neumonitis mediada inmunológicamente: Se reportó neumonitis (incluyendo los casos fatales) en pacientes que recibieron KEYTRUDA® [ver Reacciones Adversas]. Monitorear a los pacientes para detectar signos y síntomas de neumonitis. Si se sospecha neumonitis, evaluar con imagenes radiográficas y excluir otras causas. Administrar corticoides para Grado 2 o eventos mayores (dosis inicial de 1-2 mg/kg/día de prednisona o su equivalente, seguida de una reducción de la dosis), suspender KEYTRUDA® en caso de neumonitis moderada (Grado 2) y descontinuar permanentemente KEYTRUDA® en neumonitis severa (Grado 3), con riesgo para la vida (Grado 4) o moderada recurrente (Grado 2). [Ver Dosis y administración y Reacciones adversas mediadas inmunológicamente mencionadas anteriormente.]
Colitis mediada inmunológicamente: Se ha reportado colitis en pacientes que reciben KEYTRUDA® [ver Reacciones adversas]. Monitorear a los pacientes para detectar signos y síntomas de colitis y excluir otras causas. Administrar corticoides para los eventos Grado 2 o mayores (dosis inicial de 1-2 mg/kg/día de prednisona o su equivalente, seguida de una reducción de la dosis), suspender KEYTRUDA® en caso de colitis moderada (Grado 2) o colitis severa (Grado 3) y descontinuar permanentemente KEYTRUDA® en caso de colitis que ponga en riesgo la vida (Grado 4). [Ver Dosis y administración y las Reacciones adversas mediadas inmunológicamente mencionadas anteriormente.]
Hepatitis mediada inmunológicamente: Se ha reportado hepatitis en pacientes que reciben KEYTRUDA® [ver Reacciones adversas]. Monitorear a los pacientes para detectar cambios en la función hepática (al inicio del tratamiento, periódicamente durante el tratamiento y como se indica con base en la evaluación clínica) y síntomas de hepatitis y excluir otras causas. Administrar corticoides (dosis inicial 0.5-1 mg/kg/día [para eventos Grado 2] y 1-2 mg/kg/día [para eventos Grado 3 o mayores] prednisona o su equivalente, seguido de una reducción de la dosis) y con base en la severidad de las elevaciones de las enzimas hepáticas, interrumpir o suspender KEYTRUDA®. [Ver Dosis y administración y Reacciones adversas mediadas inmunológicamente mencionadas anteriormente.]
Nefritis mediada inmunológicamente: Se ha reportado nefritis en pacientes que reciben KEYTRUDA® [ver Reacciones adversas]. Monitorear a los pacientes para detectar cambios en la función renal y excluir otras causas. Administrar corticoides para eventos Grado 2 o mayores (dosis inicial de 1-2 mg/kg/día de prednisona o su equivalente, seguida de una reducción de la dosis), suspender KEYTRUDA® en caso de nefritis moderada (Grado 2) y descontinuar permanentemente KEYTRUDA® en caso de nefritis severa (Grado 3) o que ponga en riesgo la vida (Grado 4). [Ver Dosis y administración y Reacciones adversas mediadas inmunológicamente mencionadas anteriormente.]
Endocrinopatías mediadas inmunológicamente: Se ha reportado hipofisitis en pacientes que reciben KEYTRUDA® [ver Reacciones adversas].
Monitorear a los pacientes para detectar signos y síntomas de hipofisitis (incluyendo hipopituitarismo e insuficiencia suprarrenal secundaria) y excluir otras causas. Administrar corticoides para tratar la insuficiencia suprarrenal secundaria y hacer reemplazo hormonal según lo indicado clínicamente, suspender KEYTRUDA® en caso de hipofisitis moderada (Grado 2), interrumpir o suspender KEYTRUDA® en caso de hipofisitis severa (Grado 3) o que ponga en riesgo la vida (Grado 4). [Ver Dosis y administración y Reacciones adversas mediadas inmunológicamente mencionadas anteriormente.]
Se ha reportado diabetes mellitus tipo 1, incluyendo cetoacidosis diabética, en pacientes que reciben KEYTRUDA® [ver Reacciones adversas]. Monitorear a los pacientes para detectar hiperglucemia u otros signos y síntomas de diabetes. Administrar insulina para la diabetes tipo 1 y suspender KEYTRUDA® en casos de hiperglucemia severa, hasta que se logre el control metabólico.
Se han reportado trastornos de la tiroides en pacientes que reciben KEYTRUDA® y pueden ocurrir en cualquier momento durante el tratamiento; por tanto, se debe monitorear a los pacientes para detectar cambios en la función tiroidea (al inicio del tratamiento, periódicamente durante el tratamiento y según lo indicado en base a la evaluación clínica) y signos y síntomas clínicos de trastornos de la tiroides. El hipotiroidismo se puede manejar con terapia de reemplazo sin interrupción del tratamiento y sin corticoides. El hipertiroidismo puede manejarse sintomáticamente. Interrumpir o suspender KEYTRUDA® en caso de hipertiroidismo severo (Grado 3) o que ponga en riesgo la vida (Grado 4). [Ver Dosis y administración, Reacciones adversas y Reacciones adversas mediadas inmunológicamente mencionadas anteriormente.]
Se puede considerar la continuación de KEYTRUDA® en pacientes con endocrinopatía severa (Grado 3) o que ponga en riesgo la vida (Grado 4) que mejoran a Grado 2 o menor y se controlan con reemplazo hormonal.
Otras reacciones adversas mediadas inmunológicamente: Las siguientes reacciones adversas adicionales mediadas inmunológicamente, clínicamente significativas, fueron reportadas en menos del 1% (a menos que se indique lo contrario) de los pacientes tratados con KEYTRUDA® en KEYNOTE-001, KEYNOTE-002, KEYNOTE-006 y KEYNOTE-010: uveítis, miositis, síndrome Guillain-Barré, pancreatitis y reacciones cutáneas severas (1.4%).
Casos de estas reacciones adversas inmunomediadas, algunas de las cuales fueron severas, han sido reportadas en estudios clínicos o en uso post-comercialización.
Reacciones relacionadas con la infusión: Se han reportado reacciones severas a la infusión en 6 (0.2%) de los 2799 pacientes que reciben KEYTRUDA® en KEYNOTE-001, KEYNOTE-002, KEYNOTE-006 y KEYNOTE-010. En caso de reacciones severas a la infusión, suspender la infusión y descontinuar permanentemente KEYTRUDA® [ver Dosis y administración].
Los pacientes con reacción leve o moderada a la infusión pueden continuar recibiendo KEYTRUDA® con supervisión cercana; se puede considerar la premedicación con antipiréticos y antihistamínicos.
DOSIS Y ADMINISTRACIÓN:
General:
Selección de pacientes:
Carcinoma de pulmón de células no pequeñas: Los pacientes para tratamiento de NSCLC avanzado con KEYTRUDA®, deben ser seleccionados en base a la presencia de expresión positiva de PD-L1 [ver Estudios clínicos].
Dosis recomendada: KEYTRUDA® es administrado como una infusión intravenosa durante 30 minutos cada 3 semanas.
La dosis recomendada de KEYTRUDA® es:
• 200 mg para NSCLC no tratado previamente.
• 2 mg/kg para melanoma o NSCLC previamente tratado.
Los pacientes deben ser tratados con KEYTRUDA® hasta la progresión de la enfermedad o presencia de toxicidad inaceptable. Se han observado respuestas atípicas (es decir, un aumento inicial y transitorio en el tamaño del tumor o nuevas lesiones pequeñas dentro de los primeros meses, seguidas de contracción del tumor). Los pacientes clínicamente estables, con evidencia inicial de progresión de la enfermedad, deben permanecer en tratamiento hasta que se confirme la progresión de la enfermedad.
Modificaciones de la dosis:
Interrumpir KEYTRUDA® por reacciones adversas mediadas inmunológicamente, incluyendo [ver Advertencias y precauciones]:
• Neumonitis–moderada (Grado 2; National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI CTCAE v.4)).
• Colitis–moderada o severa (Grado 2 o 3).
• Nefritis–moderada (Grado 2).
• Endocrinopatías–severas o potencialmente mortales (Grado 3 o 4).
• Hepatitis asociada con:
- Elevación de la aspartato aminotransferasa (AST) o de la alanina aminotransferasa (ALT) >3 a 5 veces el límite superior normal (LSN) o bilirrubina total >1.5 a 3 veces el LSN.
Reanudar KEYTRUDA® en pacientes cuyas reacciones adversas se recuperen a Grado 0-1.
Suspender permanentemente KEYTRUDA® [ver Advertencias y precauciones (4)]:
• Si la dosis del corticoide no puede reducirse a ≤10 mg de prednisona o su equivalente por día, durante 12 semanas.
• Si la toxicidad relacionada con el tratamiento no se resuelve a Grado 0-1, en las 12 semanas siguientes a la administración de la última dosis de KEYTRUDA®.
• Si ocurre otro episodio de cualquier toxicidad severa
• Por reacciones adversas, incluyendo:
- Toxicidad que represente peligro para la vida (Grado 4), excepto endocrinopatías que mejoren a Grado 2 o menos y que se controlen con hormonas de reemplazo.
- Neumonitis mediada inmunológicamente, severa o potencialmente mortal (Grado 3 o 4) o moderada y recurrente (Grado 2).
- Nefritis mediada inmunológicamente, de intensidad severa o que compromete la vida (Grado 3 o 4).
- Hepatitis mediada inmunológicamente asociada con:
- AST o ALT >5 veces el LSN o bilirrubina total >3 veces el LSN.
- Para pacientes con metástasis hepática que inicien tratamiento con elevación moderada (Grado 2) de la AST o ALT, si la AST o la ALT aumentan ≥50% con respecto al valor basal y dura ≥1 semana.
- Reacciones severas o potencialmente mortales, relacionadas con la infusión (Grado 3 o 4).
Preparación y administración:
• Proteger de la luz. No congelar. No agitar.
• Equilibrar el vial de KEYTRUDA® a temperatura ambiente.
• Antes de la dilución, el vial del líquido puede estar fuera de refrigeración (temperatura de 25 °C o menos) hasta por 24 horas.
• Los medicamentos parenterales deben ser inspeccionados visualmente para detectar materias particuladas y decoloración antes de la administración. KEYTRUDA® es una solución transparente a ligeramente opalescente, incolora a ligeramente amarilla. Desechar el vial si se observan partículas visibles.
• Retirar el volumen requerido hasta 4 mL (100 mg) de KEYTRUDA® y transferir a una bolsa intravenosa que contenga cloruro de sodio al 0.9% o glucosa (dextrosa) al 5%, para preparar una solución diluida con una concentración final que oscile de 1 a 10 mg/mL. Mezclar la solución diluida mediante inversión suave.
• No congelar la solución para infusión.
• El producto no contiene conservantes. El medicamento diluido debe utilizarse inmediatamente. Si no se utiliza inmediatamente, las soluciones diluidas de la solución de KEYTRUDA® se pueden conservar a temperatura ambiente por un periodo acumulativo de hasta 6 horas. Las soluciones diluidas de KEYTRUDA® también se pueden conservar en refrigeración a una temperatura entre 2 °C y 8 °C; sin embargo, el tiempo total desde la dilución de KEYTRUDA® hasta terminar la infusión no debe exceder 24 horas. Si se refrigera, permita que los viales y/o bolsas IV alcancen la temperatura ambiente antes de utilizarlas.
• Administrar la solución para infusión por vía intravenosa durante 30 minutos, utilizando una línea de infusión de baja unión a proteínas 0.2 a 5 µm, estéril, no pirogénica o adicionando un filtro.
• No administrar concomitantemente otros medicamentos a través de la misma línea de infusión.
• Desechar cualquier porción no utilizada del vial.
Pacientes pediátricos: Todavía no se ha establecido la seguridad, ni la eficacia de KEYTRUDA® en menores de 18 años de edad.
Pacientes geriátricos: No se reportaron diferencias generales, en la seguridad o eficacia, entre pacientes de edad avanzada (65 años y más) y pacientes más jóvenes (menos de 65 años). No fue necesario ningún ajuste de dosis en esta población.
Insuficiencia renal: No es necesario hacer ajuste de dosis para pacientes con insuficiencia renal leve o moderada. KEYTRUDA® no ha sido estudiado en pacientes con insuficiencia renal severa.
Insuficiencia hepática: No es necesario hacer ajuste de dosis en pacientes con insuficiencia hepática leve. KEYTRUDA® no ha sido estudiado en pacientes con insuficiencia hepática moderada o severa.
SOBREDOSIS: No hay información sobre sobredosificación con KEYTRUDA®. No se ha determinado la dosis máxima tolerada de KEYTRUDA®. En ensayos clínicos, los pacientes recibieron hasta 10 mg/kg con un perfil de seguridad similar al observado en pacientes que recibieron 2 mg/kg.
En caso de sobredosis, los pacientes deben ser vigilados estrechamente en busca de signos o síntomas de reacciones adversas y deben recibir tratamiento sintomático apropiado.
PRESENTACIÓN: Caja con un vial con 4 mL de solución inyectable. (Reg. San. INVIMA 2017M – 0017599)
Fecha de revisión: Este documento fue revisado por última vez en Octubre de 2016.
LPI-MK3475-IV-102016.
MERCK SHARP & DOHME
Bogotá, D.C. - Colombia
ALMACENAMIENTO: Conservar en refrigeración a una temperatura entre 2 °C y 8 °C (36 °F y 46 °F). Proteger de la luz. No congelar. No agitar.
Para las condiciones de almacenamiento después de la dilución del medicamento, ver Dosis y Administración.


