

LYXUMIA 10 MCG / LYXUMIA 20 MCG
LIXISENATIDA
Solución inyectable
Caja , Pluma prellenada , 10 mcg
Caja , 1, 2 y 6 Pluma prellenada , 20 mcg
Caja , Pluma prellenada , 10 mcg
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN
LYXUMIA® 10 mcg: Cada ml contiene 50 mcg de lixisenatida. Cada dosis (0,2 ml) contiene 10 mcg de lixisenatida.
Vehículo/excipientes: Glicerol 85%, acetato de sodio trihidratado, metionina, metacresol, solución de ácido clorhídrico/ hidróxido de sodio para ajuste de pH, agua para inyección c.s.p. 1 ml.
LYXUMIA® 20 mcg: Cada ml contiene 100 mcg de lixisenatida. Cada dosis (0,2 ml) contiene 20 mcg de lixisenatida.
Vehículo/excipientes: Glicerol 85%, acetato de sodio trihidratado, metionina, metacresol, solución de ácido clorhídrico/ hidróxido de sodio para ajuste de pH, agua para inyección c.s.p 1 ml.
Vía de administración: Subcutánea
Industria Alemana
VENTA BAJO RECETA MÉDICA
INDICACIONES: LYXUMIA® está indicado para el tratamiento de adultos con diabetes mellitus tipo 2 para lograr el control glucémico en pacientes que no están controlados con la terapia estándar, de la siguiente forma:
• En combinación con los siguientes antidiabéticos orales: Metformina, una sulfonilurea o una combinación de estos agentes.
• En combinación con insulina basal: Sola, en combinación con metformina o en combinación con una sulfonilurea.
ACCIÓN TERAPÉUTICA: Medicamento antidiabético - reductor de la glucosa en sangre. Código ATC:A10BX10.
CARACTERÍSTICAS FARMACOLÓGICAS / PROPIEDADES
Grupo farmacoterapéutico: Otros fármacos reductores de la glucosa en sangre, excluyendo insulinas.
FARMACOCINÉTICA
Absorción: Después de la administración subcutánea a pacientes con diabetes tipo 2, la velocidad de absorción de lixisenatida es rápida y no está influenciada por la dosis administrada. Independientemente de la dosis y la administración de lixisenatida como dosis única o múltiple, la mediana del Tmáx es de 1 a 3,5 horas en pacientes con diabetes tipo 2. No hay diferencias clínicamente relevantes en la velocidad de absorción cuando lixisenatida se administra por vía subcutánea en abdomen, muslo o brazo.
Distribución: Lixisenatida tiene un nivel moderado de unión a las proteínas humanas (55%).
El volumen de distribución después de la administración subcutánea de lixisenatida en pacientes con diabetes tipo 2 varió entre 90 y 140 L después de una sola administración y entre 90 y 120 L en estado estacionario, independientemente de la dosis administrada.
Metabolismo: Como péptido, lixisenatida se elimina a través de filtración glomerular, seguida por reabsorción tubular y la subsecuente degradación metabólica, lo que resulta en péptidos y aminoácidos de menor tamaño que son reintroducidos en el metabolismo de las proteínas.
Eliminación: Después de la administración de dosis múltiples en pacientes con diabetes tipo 2, el promedio de la vida media de eliminación aparente generalmente varía de 1,5 a 4,5 horas y el promedio de la depuración aparente varía de 20 a 67 l/h en estado estacionario.
Poblaciones especiales
Género: El género no afecta la farmacocinética de lixisenatida, en base al análisis de los datos de la farmacocinética de la población.
Personas de edad avanzada: La edad no tiene un efecto clínicamente relevante en la farmacocinética de lixisenatida, en base al análisis de los datos de la farmacocinética de la población en pacientes con diabetes tipo 2 y en un estudio de farmacocinética realizado en sujetos de edad avanzada no diabéticos.
Raza: El origen étnico no tuvo efecto clínicamente relevante en la farmacocinética de lixisenatida, en base a los resultados de los estudios de farmacocinética en sujetos caucásicos, japoneses y chinos y en base a un análisis de los datos de la farmacocinética de la población que incluyó pacientes caucásicos y asiáticos (japoneses).
Pacientes con deterioro hepático: Debido a que lixisenatida es depurada principalmente por el riñón no se han realizado estudios de farmacocinética en pacientes con deterioro hepático agudo o crónico. No se espera que la disfunción hepática afecte la farmacocinética de lixisenatida.
Pacientes con deterioro renal: No hubo diferencias relevantes en la media de la depuración, la Cmáx y el Área Bajo la Curva (ABC) de lixisenatida entre los sujetos con función renal normal y los sujetos con deterioro leve o moderado de la función renal. La media de la Cmáx y del ABC incrementó con el aumento del grado de deterioro renal.
FARMACODINÁMICA
Mecanismo de acción: Lixisenatida es un agonista potente y selectivo del receptor del GLP-1. El receptor del GLP-1 es el ligando para el GLP-1 natural, una hormona endógena del grupo de las incretinas que potencia la secreción de insulina glucosa-dependiente de las células beta pancreáticas.
La acción de lixisenatida es mediada a través de una interacción específica con los receptores de GLP-1, lo que conduce a un aumento de monofosfato de adenosina cíclico (AMPc) intracelular. Lixisenatida estimula la secreción de insulina cuando se encuentra aumentada la glucosa en sangre pero no durante la normoglucemia, lo cual limita el riesgo de hipoglucemia. Al mismo tiempo, se suprime la secreción de glucagón. En caso de hipoglucemia el mecanismo de rescate de secreción de glucagón se conserva. Lixisenatida mostró además una tendencia hacia la actividad insulinotrópica, incluyendo mejoramiento de la biosíntesis de insulina y estimulación de la proliferación de células beta en animales.
Lixisenatida hace más lento el vaciado gástrico, reduciendo con ello la velocidad a la cual aparece en la circulación la glucosa derivada de los alimentos. El efecto en el vaciado gástrico también podría contribuir a reducir el peso corporal.
Efectos farmacodinámicos: Cuando se administra una vez al día, lixisenatida mejora el control glucémico por medio de los efectos reductores inmediatos y sostenidos de las concentraciones tanto postprandial como en ayunas en pacientes con diabetes tipo 2.
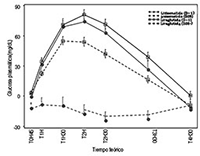
Este efecto en la glucosa post-prandial se confirmó en un estudio a 4 semanas versus 1,8 mg de liraglutida una vez al día. En comparación con liraglutida, una dosis de 20 mcg de lixisenatida una vez al día demostró reducción superior del área bajo la curva de la glucemia post-prandial después de una prueba con alimento. (ver Figura 1).
Figura 1: Media (± SEM) corregida del perfil de glucemia post-prandial el Día -1 y el Día 28, por tratamiento
Eficacia clínica/ Estudios clínicos: Los efectos de LYXUMIA® en el control glucémico se evaluaron en seis estudios clínicos aleatorios, doble ciego, controlados con placebo y un estudio aleatorio, abierto, controlado con activo versus exenatida.
Estos estudios incluyeron 3825 pacientes con diabetes tipo 2 (2445 pacientes aleatorizados a lixisenatida), 48,2% de hombres y 51,8% de mujeres.
768 sujetos (447 aleatorizados a lixisenatida) tenían = 65 años de edad y 103 sujetos (57 aleatorizados a lixisenatida) tenían =75 años de edad.
En los estudios de Fase III completados, se observó que más de 90% de la población de pacientes pudo permanecer con la dosis de mantenimiento de 20 mcg LYXUMIA® una vez al día al final del periodo de tratamiento (24 semanas).
Control glucémico: LYXUMIA® demostró un efecto superior, en comparación con placebo, para reducir la hemoglobina glucosilada (HbA1c) independientemente del tratamiento de base, y LYXUMIA® una vez al día mostró reducción no inferior de la HbA1c en comparación con exenatida dos veces al día.
Este efecto en la HbA1c se mantuvo hasta por 2 años en los estudios a largo plazo.
La reducción de la HbA1c fue significativa con la administración una vez al día en la mañana o en la tarde.
Tratamiento de combinación complementario con antidiabéticos orales: LYXUMIA® en combinación con metformina, una sulfonilurea o una combinación de estos agentes, mostró reducciones clínica y estadísticamente significativas en la HbA1c, en la glucosa plasmática en ayunas y en la glucosa post-prandial de 2 horas después de una prueba con alimento, en comparación con placebo al final del periodo principal de tratamiento de 24 semanas. (Tablas 1 y 2).
Tratamiento complementario a metformina sola
|
Tabla 1. Estudios controlados con placebo en combinación con metformina (resultados de 24 semanas). |
||||||
|
Metformina como tratamiento de base |
||||||
|
Lixisenatida 20 mcg |
Placebo (N= 159) |
Lixisenatida 20 mcg |
Placebo (N= 170) |
|||
|
Inicio de la dosis en dos pasos* (N= 160) |
Inicio de la dosis en un paso* (N= 160) |
Mañana (N= 255) |
Tarde (N= 255) |
|||
|
Media de HbA1c (%) Línea basal Cambio en la media de cuadrados mínimos respecto a la línea basal |
8,12 |
7,99 |
8,03 |
8,07 |
8,07 |
8,02 |
|
-0,83 |
-0,92 |
-0,42 |
-0,87 |
-0,75 |
-0,38 |
|
|
Pacientes (%) que alcanzaron HbA1c < 7.0% |
42,1 |
47,4 |
24,1 |
43,0 |
40,6 |
22,0 |
|
Media del peso corporal (kg) Línea basal Cambio en la media de cuadrados mínimos respecto a la línea basal |
88,08 |
90,30 |
87,86 |
90,14 |
89,01 |
90,40 |
|
-2,68 |
-2,63 |
-1,63 |
-2,01 |
-2,02 |
-1,64 |
|
|
*En este estudio se evaluaron dos regímenes de inicio de dosis de 2 semanas de duración; ambos fueron seguidos por un periodo de mantenimiento con 20 mcg de LYXUMIA® una vez al día. El inicio en un paso (10 mcg por dos semanas) seguido por 20 mcg para mantenimiento, es el régimen recomendado para uso. |
||||||
En el estudio con control activo, LYXUMIA® una vez al día mostró reducción no inferior de HbA1c en comparación con exenatida dos veces al día al final del periodo principal de tratamiento de 24 semanas (respectivamente -0,79% y -0,96%) y un porcentaje similar de pacientes que alcanzaron una HbA1c menor de 7% en el grupo de LYXUMIA® (48,5%) y en el grupo de exenatida (49,8%).
Tratamiento complementario a una sulfonilurea sola o en combinación con metformina
|
Tabla 2. Estudio controlado con placebo en combinación con una sulfonilurea (resultados de 24 semanas) |
||
|
Sulfonilurea como tratamiento de base con o sin metformina |
||
|
Lixisenatida 20 mcg (N= 570) |
Placebo (N= 286) |
|
|
Media de HbA1c (%) Línea basal |
8,28 |
8,22 |
|
Cambio en la media de cuadrados mínimos respecto a la línea basal |
-0,85 |
-0,10 |
|
Pacientes (%) que alcanzaron HbA1c < 7,0% |
36,4 |
13,5 |
|
Media del peso corporal (kg) Línea basal |
82,58 |
84,52 |
|
Cambio en la media de cuadrados mínimos respecto a la línea basal |
-1,76 |
-0,93 |
Tratamiento de combinación complementario con una insulina basal: En comparación con placebo, LYXUMIA® administrado con una insulina basal sola, o con una combinación de una insulina basal y metformina, o una combinación de una insulina basal y una sulfonilurea, produjo reducciones estadísticamente significativas de la HbA1c y de la glucosa post-prandial de 2 horas después de una prueba con alimento. Al final del periodo principal de tratamiento de 24 semanas, la reducción en la dosis diaria de insulina desde la línea basal fue mayor en el grupo de LYXUMIA® que en el grupo de placebo.
|
Tabla 3. Estudio controlado con placebo en combinación con una insulina basal (resultados de 24 semanas) |
||||
|
Insulina basal como tratamiento de base |
Insulina basal como tratamiento de base |
|||
|
Lixisenatida 20 mcg (N= 327) |
Placebo (N= 166) |
Lixisenatida 20 mcg (N= 154) |
Placebo (N= 157) |
|
|
Media de HbA1c (%) Línea basal |
8,39 |
8,38 |
8,53 |
8,53 |
|
Cambio en la media de cuadrados mínimos respecto a la línea basal |
-0,74 |
-0,38 |
-0,77 |
0,11 |
|
Pacientes (%) que alcanzaron HbA1c < 7,0% |
28,3 |
12,0 |
35,6 |
5,2 |
|
Media del cambio en la dosis de insulina basal (U) Línea basal |
53,62 |
57,65 |
24,87 |
24,11 |
|
Cambio en la media de cuadrados mínimos respecto a la línea basal |
-5,62 |
-1,93 |
-1,39 |
-0,11 |
|
Media del peso corporal (kg) Línea basal |
87,39 |
89,11 |
65,99 |
65,60 |
|
Cambio en la media de cuadrados mínimos respecto a la línea basal |
-1,80 |
-0,52 |
-0,38 |
0,06 |
Glucosa plasmática en ayunas: En los estudios controlados con placebo, las reducciones en la glucosa plasmática en ayunas obtenidas con el tratamiento con LYXUMIA® variaron desde 0,42 mmol/l hasta 1,19 mmol/l al final del periodo principal de tratamiento de 24 semanas.
Glucosa post-prandial: El tratamiento con LYXUMIA® produjo, en comparación con placebo, reducciones estadísticamente superiores en la glucosa post-prandial de 2 horas después de una prueba con alimento independientemente del tratamiento de base.
Las reducciones con LYXUMIA® variaron desde 4,51 hasta 7,96 mmol/l de la línea basal al final del periodo principal de tratamiento de 24 semanas a través de todos los estudios en los que se midió la glucosa post-prandial; 26,2% a 46,8% de pacientes tuvieron un valor de glucosa post-prandial de 2 horas menor de 7,8 mmol/l (140,54 mg/dl).
Peso corporal: El tratamiento con LYXUMIA® en combinación con metformina, una insulina basal y/o una sulfonilurea produjo una reducción en la media del peso corporal de hasta 2,96 kg al final del periodo principal de tratamiento de 24 semanas el cual se mantuvo en los estudios a largo plazo hasta por 2 años.
La reducción del peso corporal es independiente de la ocurrencia de náusea y vómito.
Función de las células beta: En estudios clínicos, LYXUMIA® mejoró la función de las células beta por medio de evaluación de la función de las células beta con el modelo homeostático (HOMA-ß).
La restauración de la secreción de insulina en la primera fase y la mejoría en la secreción de insulina en la segunda fase en respuesta a un bolo intravenoso de glucosa, se demostró en pacientes con diabetes tipo 2 (n=20) después de una dosis única de LYXUMIA®.
Frecuencia cardiaca: No se observó aumento de la frecuencia cardiaca en todos los estudios de fase III controlados.
En un estudio a 4 semanas versus liraglutida, la media de la frecuencia cardiaca disminuyó en 3,6 latidos/min en el grupo de lixisenatida (20 mcg una vez al día) mientras que aumentó en 5,3 latidos/min en el grupo de liraglutida (1,8 mg una vez al día).
Presión arterial: Se observaron reducciones de la presión arterial sistólica y diastólica de hasta 2,1 mmHg y hasta 1,5 mmHg, respectivamente, en los estudios de fase III controlados con placebo.
CARCINOGENICIDAD: En estudios de carcinogenicidad a 2 años (uso subcutáneo) no se observó carcinoma de células C en ningún nivel de dosis en ratones, y el nivel sin efecto (NOEL o No Effect Level) para carcinomas de células C fue de 40 mcg/kg dos veces al día en ratas. Se observaron efectos proliferativos en células C de la tiroides en ratas y ratones a índices de exposición muy altos (respectivamente =913 veces y =272 veces) en comparación con la exposición en humanos en la dosis terapéutica. Se considera que estos hallazgos son causados por un mecanismo mediado por el receptor de GLP-1 al cual los roedores son particularmente sensibles.
CONTRAINDICACIONES: LYXUMIA® es contraindicada en pacientes con hipersensibilidad conocida a la sustancia activa, lixisenatida, o a cualquiera de los excipientes de la formulación.
USO EN EL EMBARAZO Y LACTANCIA: No hay datos adecuados del uso de LYXUMIA® en mujeres embarazadas. Los estudios en animales han mostrado toxicidad reproductiva (ver adelante). Se desconoce el potencial riesgo para los humanos. LYXUMIA® no debe utilizarse durante el embarazo, y en su lugar se recomienda el uso de insulina. Si una paciente desea embarazarse, o si ocurre un embarazo, el tratamiento con LYXUMIA® debe descontinuarse.
Se desconoce si LYXUMIA® se excreta en la leche humana. Debido a la falta de experiencia, LYXUMIA® no debe utilizarse durante la lactancia.
CAPACIDAD PARA CONDUCIR AUTOS Y UTILIZAR MAQUINARIA: No se han llevado a cabo estudios sobre los efectos en la capacidad para conducir autos y utilizar maquinaria.
Cuando se utiliza en combinación con una sulfonilurea o una insulina basal, se debe aconsejar a los pacientes para que tomen precauciones para evitar la hipoglucemia mientras conducen o utilizan maquinaria.
REACCIONES ADVERSAS: Las frecuencias de reacciones adversas se definen como: Muy común: = 10%; común: = 1 a < 10%; poco común: = 0,1 a < 1%; rara: = 0,01% a < 1%; muy rara: < 0,01%), desconocida (no puede ser estimada a partir de datos disponibles). Más de 2600 pacientes han recibido LYXUMIA® ya sea solo o en combinación con metformina, una sulfonilurea (con o sin metformina) o una insulina basal (con o sin metformina, o con o sin una sulfonilurea) en 8 estudios de fase III a gran escala controlados con placebo o con control activo.
Las reacciones adversas reportadas con más frecuencia durante los estudios clínicos fueron náuseas y vómitos. Estas reacciones en su mayoría fueron leves y transitorias.
En la Tabla 4 se listan las reacciones adversas reportadas durante el periodo total de tratamiento en estudios de fase III controlados con placebo y con control activo. En la tabla se presentan las reacciones adversas por término preferente que ocurrieron con una incidencia >5% cuando la frecuencia fue mayor entre los pacientes tratados con LYXUMIA® que en los pacientes tratados con todos los comparadores. En la tabla también se incluyen las reacciones adversas con una frecuencia = 2% en el grupo de LYXUMIA®, cuando la frecuencia fue >2 veces la frecuencia para el grupo comparador.
|
Tabla 4. Reacciones adversas reportadas durante el periodo total de tratamiento en los estudios de fase III controlados con placebo y con control activo (incluyendo el periodo posterior al periodo principal de tratamiento de 24 semanas en estudios con =76 semanas de tratamiento total). |
||
|
Clase de sistema-órgano del MedDRA / términos de reacción adversa |
Frecuencia de ocurrencia |
|
|
Reacciones |
Muy común |
Común |
|
Infecciones e infestaciones |
||
|
Influenza |
X |
|
|
Infecciones de vías respiratorias superiores |
X |
|
|
Trastornos del metabolismo y de nutrición |
||
|
Hipoglucemia sintomática (cuanto el tratamiento incluye una sulfonilurea y/o una insulina basal) |
X |
|
|
Trastornos del sistema nervioso |
||
|
Cefalea |
X |
|
|
Mareo |
X |
|
|
Trastornos gastrointestinales |
||
|
Náusea |
X |
|
|
Diarrea |
X |
|
|
Vómito |
X |
|
|
Dispepsia |
X |
|
|
Trastornos musculoesqueléticos y del tejido conectivo |
||
|
Dolor de espalda |
X |
|
Hipoglucemia: En pacientes a los que se administró LYXUMIA® como monoterapia o en combinación con metformina sola, la hipoglucemia sintomática fue común y la tasa fue similar en los pacientes con LYXUMIA® y los pacientes con placebo durante el periodo total de tratamiento.
En pacientes a los que se administró LYXUMIA® en combinación con una sulfonilurea o una insulina basal, la hipoglucemia sintomática fue muy común.
Durante el periodo total de tratamiento, la tasa no fue sustancialmente más alta en los pacientes con LYXUMIA® que en los pacientes con placebo cuando LYXUMIA® se administró en combinación con:
• Una sulfonilurea y metformina,
• una insulina basal sola,
• una insulina basal y metformina.
Durante el periodo total de tratamiento, cuando LYXUMIA® se administró con una sulfonilurea sola, ocurrió hipoglucemia sintomática en 22,7% de los pacientes tratados con LYXUMIA® versus 15,2% de los tratados con placebo. Cuando LYXUMIA® se administró con una sulfonilurea y una insulina basal, ocurrió hipoglucemia sintomática en 47,2% de los pacientes tratados con LYXUMIA® en comparación con 21,6% de los tratados con placebo.
En general, la incidencia de hipoglucemia sintomática severa fue poco común (0,4% en los pacientes con LYXUMIA® y 0,2% en los pacientes con placebo) durante el periodo total de tratamiento de los estudios de Fase III controlados con placebo.
Trastornos gastrointestinales: Náuseas y vómitos fueron las reacciones adversas reportadas con más frecuencia durante el periodo principal de tratamiento de 24 semanas. La incidencia de náuseas fue mayor en el grupo de LYXUMIA® (26,1%) en comparación con el grupo de placebo (6,2%) y la incidencia de vómito fue mayor en el grupo de LYXUMIA® (10,5%) que en el de placebo (1,8%). Las reacciones en su mayoría fueron leves y transitorias y ocurrieron durante las primeras 3 semanas después de iniciar el tratamiento. Después, disminuyeron progresivamente durante las siguientes semanas.
La incidencia de náuseas fue más baja en el grupo de LYXUMIA® (24,5%) en comparación con el grupo de exenatida dos veces al día (35,1%) y fue similar para los otros eventos gastrointestinales.
Reacciones en el sitio de la inyección: Las reacciones en el sitio de la inyección se han reportado en 3,9% de los pacientes que recibieron LYXUMIA® mientras que se reportaron en 1,4% de los pacientes que recibieron placebo durante el periodo principal de tratamiento de 24 semanas. La mayoría de las reacciones fueron de intensidad leve y generalmente no causaron la descontinuación del tratamiento.
Inmunogenicidad: Consistentemente con las propiedades potencialmente inmunogénicas de los productos medicinales que contienen proteínas o péptidos, los pacientes pueden desarrollar anticuerpos anti-lixisenatida después del tratamiento con LYXUMIA® y, al final del periodo principal de tratamiento (de 24 semanas) en estudios controlados con placebo, 69,4% de los pacientes con lixisenatida tuvieron un estado positivo para anticuerpo. Sin embargo, el cambio en la HbA1c desde la línea basal fue similar independientemente del estado de anticuerpo (positivo o negativo).
De los pacientes tratados con lixisenatida, el 79,3% tuvo un estado de anticuerpo negativo o una concentración de anticuerpo por debajo del límite inferior de cuantificación. El otro 20,7% de los pacientes tuvo una concentración de anticuerpo cuantificada y algunos de estos pacientes tuvieron disminución de la eficacia asociada con una concentración elevada de anticuerpos anti-lixisenatida.
No hubo diferencia en el perfil global de seguridad en los pacientes independientemente del estado de anticuerpo, con excepción de un aumento en la incidencia de reacciones en el sitio de la inyección para los pacientes positivos para anticuerpo. La mayoría de las reacciones en el sitio de la inyección fueron leves, independientemente del estado de anticuerpo.
No hubo reactividad cruzada versus glucagón natural o GLP-1 endógeno.
Reacciones alérgicas: Las reacciones alérgicas (como reacción anafiláctica, angioedema y urticaria) se han reportado en 0,4% de los pacientes con LYXUMIA® en comparación con menos de 0,1% en pacientes con placebo durante el periodo principal de tratamiento de 24 semanas.
Descontinuación del medicamento: La incidencia de descontinuación del tratamiento debido a eventos adversos fue del 7,4% para LYXUMIA® en comparación con 3,2% en el grupo de placebo durante el periodo principal de tratamiento de 24 semanas. Los eventos adversos más comunes que condujeron a descontinuación del tratamiento en el grupo de LYXUMIA® fueron náusea (3,1%) y vómito (1,2%).
INTERACCIONES MEDICAMENTOSAS: Lixisenatida es un péptido y no es metabolizado por el citocromo P450. En estudios in vitro, lixisenatida no afectó la actividad de las isoenzimas del citocromo P450 o los transportadores humanos sometidos a prueba.
El retraso del vaciado gástrico con lixisenatida puede influir en la absorción de los medicamentos administrados por vía oral. Para medicamentos orales que son particularmente dependientes de las concentraciones umbral para eficacia, debe advertírse a los pacientes que tomen esos medicamentos al menos 1 hora antes u 4 horas después de la inyección de lixisenatida.
Paracetamol: Después de la administración de una dosis única de 1000 mg de paracetamol, el ABC y el t1/2 de paracetamol permanecieron sin cambio sin importar la hora en que fue administrado (antes o después de la inyección de lixisenatida). Cuando se administró 1 o 4 horas después de la inyección de lixisenatida, la Cmáx de paracetamol disminuyó 29% y 31%, respectivamente, y la mediana del tmáx se retrasó 2,0 y 1,75 horas, respectivamente.
Con base en estos resultados, no se requiere ajuste de la dosis de paracetamol.
Anticonceptivos orales: Después de la administración de una dosis única de un anticonceptivo oral (etinilestradiol 0,03 mg /levonorgestrel 0,15 mg) 1 hora antes u 11 horas después de la inyección subcutánea de lixisenatida, la Cmáx, el ABC, el t1/2 y el tmáx de etinilestradiol y levonorgestrel permanecieron inalterados.
La administración de etinilestradiol y levonorgestrel 1 hora o 4 horas después de la inyección subcutánea de lixisenatida no afectó el ABC y el t1/2 mientras que la Cmáx de etinilestradiol disminuyó en 52% y 39%, respectivamente, y la Cmáx de levonorgestrel disminuyó en 46 y 20% respectivamente, y la mediana del tmax se retrasó 1 a 3 horas.
La reducción de la Cmáx es de relevancia clínica limitada y no se requiere ajuste de la dosis para los anticonceptivos orales.
Atorvastatina: Cuando lixisenatida y 40 mg de atorvastatina se coadministraron en la mañana por 6 días, la exposición de atorvastatina no fue afectada, mientras que la Cmáx disminuyó en 31% y el tmáx retrasó en 3,25 horas.
No se observó un aumento semejante para el tmáx cuando atorvastatina se administró en la tarde y lixisenatida en la mañana, pero el ABC y la Cmáx de atorvastatina aumentaron en 27% y 66%, respectivamente.
Estos cambios no son clínicamente relevantes y, por lo tanto, no se requiere ajuste de la dosis de atorvastatina cuando se coadministra con lixisenatida.
Warfarina y otros derivados de cumarina: Después de la administración concomitante de 25 mg de warfarina con lixisenatida, no hubo efectos en el ABC o el INR (Proporción Internacional Normalizada o International Normalized Ratio), mientras que la Cmáx fue reducida y el tmáx se retrasó 7 horas.
Con base en estos resultados, no se requiere ajuste de la dosis de warfarina cuando se coadministra con lixisenatida.
Digoxina: Después de la administración concomitante de lixisenatida y 0,25 mg de digoxina, el ABC de digoxina no se vio afectada. El tmáx de digoxina se retrasó 1,5 horas y la Cmáx se redujo 26%.
Con base en estos resultados, no se requiere ajuste de la dosis de digoxina cuando se coadministra con lixisenatida.
Ramipril: Después de la administración concomitante de lixisenatida y 5 mg de ramipril durante 6 días, el ABC de ramipril aumentó en 21% mientras que la Cmáx disminuyó en 63%.El ABC y la Cmáx del metabolito activo (ramiprilato) no fueron afectados. El tmáx de ramipril y ramiprilato se retrasaran en aproximadamente 2,5 horas.
Con base en estos resultados, no se requiere ajuste de la dosis de ramipril cuando se coadministra con lixisenatida.
ADVERTENCIAS
Uso en diabetes tipo 1: No hay experiencia terapéutica con LYXUMIA® en pacientes con diabetes mellitus tipo 1 y no debe utilizarse en estos pacientes. LYXUMIA® no debe usarse para el tratamiento de la cetoacidosis diabética.
Riesgo de pancreatitis: El uso de agonistas del receptor del péptido similar al glucagón tipo 1 (GLP-1 o Glucagon-like-peptide-1) ha estado asociado al riesgo de desarrollar pancreatitis aguda. Los pacientes deben ser informados de los síntomas característicos de la pancreatitis aguda: Dolor abdominal persistente y severo. Si se sospecha pancreatitis, LYXUMIA® debe descontinuarse; si se confirma pancreatitis aguda, LYXUMIA® no debe reiniciarse. Utilícese con precaución en pacientes con antecedente de pancreatitis.
PRECAUCIONES: Uso en pacientes con gastroparesia severa. El uso de agonistas del receptor de GLP-1 puede estar asociado con reacciones adversas gastrointestinales. LYXUMIA® no se ha estudiado en pacientes con enfermedad gastrointestinal severa, incluyendo gastroparesia severa y, por lo tanto, el uso de LYXUMIA® no se recomienda en estos pacientes.
Riesgo de hipoglucemia: Los pacientes que reciben LYXUMIA® con una sulfonilurea o con una combinación de una insulina basal y una sulfonilurea pueden tener un mayor riesgo de hipoglucemia. Se puede considerar reducir la dosis de la sulfonilurea o de la insulina basal para disminuir el riesgo de hipoglucemia. (ver Posología y modo de uso).
El médico deberá valorar de manera individualizada cada caso, de forma tal que determine los factores que puedan estar influyendo en el descontrol (mediante niveles de hemoglobina glucosilada) y decida la modificación del tratamiento en base al análisis realizado.
POSOLOGÍA Y MODO DE USO
Posología: La dosis inicial es de 10 mcg de LYXUMIA® una vez al día durante 14 días.
Luego, la dosis de LYXUMIA® debe aumentarse a 20 mcg una vez al día, la cual es la dosis de mantenimiento.
Cuando LYXUMIA® se agrega al tratamiento existente con metformina, la dosis actual de metformina puede continuar sin cambios.
Cuando LYXUMIA® se agrega al tratamiento existente con una sulfonilurea o una combinación de una sulfonilurea y una insulina basal, puede considerarse una reducción de la dosis de la sulfonilurea o de la insulina basal para disminuir el riesgo de hipoglucemia (ver Precauciones).
El uso de LYXUMIA® no requiere monitoreo específico de la glucosa sanguínea. Sin embargo, cuando se utiliza en combinación con una sulfonilurea o una insulina basal, el monitoreo de la glucosa sanguínea o el automonitoreo de la glucosa sanguínea pueden llegar a ser necesarios para ajustar las dosis de la sulfonilurea o de la insulina basal.
Preparación y manejo: Inspeccione LYXUMIA® antes de cada uso. LYXUMIA® sólo debe utilizarse cuando la solución es transparente, incolora y sin partículas visibles.
LYXUMIA® no debe utilizarse si se ha congelado.
LYXUMIA® puede usarse con agujas desechables calibre 29 a 32 para lapicera/pluma. No se incluyen agujas para la pluma.
Se deben dar instrucciones al paciente para que después de utilizar la aguja la deseche de acuerdo con los requerimientos locales y para que guarde la pluma sin la aguja puesta. Esto ayuda a prevenir la contaminación y la posible obstrucción de la aguja. La pluma debe utilizarse exclusivamente para un paciente.
Cualquier producto medicinal no utilizado o material de desecho deben descartarse de acuerdo con los requerimientos locales.
Modo de uso: LYXUMIA® se administra una vez al día dentro de la hora previa a la primera comida del día o la comida de la noche.
Si una dosis de LYXUMIA® se omite, debe inyectarse dentro de la hora previa a la siguiente comida.
LYXUMIA® se debe inyectar por vía subcutánea en el muslo, el abdomen o en la parte superior del brazo. LYXUMIA® no debe administrarse por vía intravenosa o intramuscular.
Compatibilidades/Incompatibilidades: En ausencia de un estudio de compatibilidad, lixisenatida no puede mezclarse con otros productos medicinales.
Poblaciones especiales
Población pediátrica: Aún no se han evaluado la seguridad y la eficacia de LYXUMIA® en pacientes menores de 18 años de edad.
Pacientes de edad avanzada (= 65 años): No se requiere ajuste de la dosis con base en la edad. (ver Farmacocinética).
Deterioro hepático: No es necesario hacer ajuste de la dosis en pacientes con deterioro hepático. (ver Farmacocinética).
Deterioro renal: No se requiere ajuste de la dosis para pacientes con deterioro renal leve (depuración de creatinina: 50-80 ml/min). Hay experiencia limitada con deterioro renal moderado (depuración de creatinina: 30-50 ml/min) y debe tenerse precaución en esta población. No hay experiencia terapéutica en pacientes con deterioro renal severo (depuración de creatinina < 30 ml/min) o enfermedad renal en etapa terminal y, por lo tanto, no se recomienda el uso de LYXUMIA® en estas poblaciones. (ver Farmacocinética).
SOBREDOSIS
Signos y síntomas: Durante los estudios clínicos, se administraron dosis de hasta 30 mcg de lixisenatida dos veces al día a pacientes con diabetes tipo 2 en un estudio de 13 semanas. Las dosis fueron bien toleradas y únicamente se observó un aumento en la incidencia de trastornos gastrointestinales.
Manejo: En caso de sobredosis debe iniciarse tratamiento de apoyo apropiado, de acuerdo a los signos y síntomas clínicos del paciente y la dosis de LYXUMIA® debe reducirse a la dosis prescrita.
Ante la eventualidad de una sobredosificación concurrir al hospital más cercano o comunicarse con los Centros de Toxicología (Léase al final del prospecto).
TERATOGENICIDAD: Retardo en el crecimiento fetal, hallazgos esqueléticos y osificación retardada se produjeron/ evidenciaron en ratas a dosis tóxicas para la madre resultando en exposiciones = 4,6 veces la exposición media a la MRHD (Dosis máxima recomendada en humanos). En conejos, se observó una mayor incidencia de variaciones de esternón y costillas se observaron a dosis tóxicas para las madres con exposiciones = 345 veces la exposición media a la MRHD (Dosis máxima recomendada en humanos).
En el estudio de toxicidad pre-postnatal en ratas, la lixisenatida causó mortalidad ligeramente aumentada en las crías a 200 mg/kg dos veces al día, disminución del crecimiento de las crías machos, disminución ligera de succión y retraso menor en el desarrollo del crecimiento del pelo en dosis de 20 y 200 mg/kg de peso corporal dos veces al día. No se observó toxicidad funcional o de comportamiento en las crías de ratas a las cuales se administró lixisenatida en cualquier dosis.
Deterioro de la fertilidad: Lixisenatida no tuvo efectos sobre la fertilidad masculina y femenina en ratas.
MUTAGENICIDAD Y GENOTOXICIDAD: Lixisenatida no tuvo efectos genotóxicos, basado en un ensayo in vivo de micronúcleos en ratones y en ensayos in vitro: La prueba de Ames modificada con o sin activación metabólica, y el ensayo in vitro de aberración cromosómica en mamíferos en linfocitos humanos cultivados.
PRESENTACIONES
LYXUMIA® 10 mcg: Caja con 1 lapicera/pluma pre llenada descartable con 3 ml, proveyendo 14 dosis de 10 mcg (Reg. San. INVIMA 2013M-0014627).
LYXUMIA® 20 mcg: Caja con 1, 2 y 6 lapiceras/plumas pre llenadas descartable con 3 ml cada una, proveyendo 14 dosis de 20 mcg (Reg. San. INVIMA 2013M-0014608).
Venta bajo fórmula médica.
Fabricado en Alemania por: Sanofi-Aventis Deutschland GmbH
Brüningstraße 50 H500, H590, H600, 65926 Frankfurt am Main.
Imp./ Distr.: SANOFI-AVENTIS de Colombia S.A., Bogotá.
ÚLTIMA REVISIÓN: Jan/2013
CCDSv.01-LRC-30-Sept-2011
SANOFI-AVENTIS DE COLOMBIA S. A.
Transversal 23 No. 97-73, Pisos 8 y 9
Teléfono: 6214400, Fax: 7444237
Bogotá, D.C., Colombia
CONDICIONES DE CONSERVACIÓN/RECOMENDACIONES SOBRE ALMACENAMIENTO
Antes del primer uso: LYXUMIA® debe ser conservado en refrigeración entre 2 °C a 8 °C, en su embalaje original, para proteger de la luz. No congelar.
Después del primer uso: LYXUMIA® puede mantenerse a una temperatura ambiente no superior a 30 °C, por hasta 14 días. No refrigerar. No congelar. La tapa en el dispositivo tipo pluma debe ser recolocada en la misma después de cada uso con el fin de protegerlo de la luz. El dispositivo no se debe guardar con la aguja puesta.
El dispositivo debe ser descartado 14 días después de la primera apertura.
MANTENER EN SU ENVASE ORIGINAL, NO DEBE UTILIZARSE DESPUÉS DE LA FECHA DE CADUCIDAD INDICADA EN EL ENVASE.
MANTENER TODOS LOS MEDICAMENTOS FUERA DEL ALCANCE DE LOS NIÑOS.
NO USAR DURANTE EL EMBARAZO Y LA LACTANCIA.


