

POLIVY
POLATUZUMAB VEDOTINA
Polvo liofilizado estéril para infusión intravenosa
Caja, 1 Vial,
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA: POLVO compacto liofilizado suministrado en viales monodosis de 20 ml que proporcionan 140 mg de polatuzumab vedotina (20 mg/ml).
INDICACIONES TERAPÉUTICAS: Polatuzumab vedotina en combinación con bendamustina y rituximab está indicado para el tratamiento de pacientes adultos con linfoma difuso de células B grandes en recaída o refractario que hayan recibido al menos dos tratamientos previos y que no sean candidatos a un trasplante de células madre hematopoyéticas.
USO EN POBLACIONES ESPECIALES: Mujeres y hombres con posibilidad de procrear: Fecundidad: Según los resultados de estudios en animales, Polivy puede afectar a la función reproductora y la fecundidad masculinas (véase el apartado 3.3.3 Trastornos de la fecundidad).
Anticoncepción
Mujeres: Se indicará a las mujeres con posibilidad de procrear que deben utilizar un método anticonceptivo eficaz durante el tratamiento con Polivy y hasta que hayan pasado al menos 9 meses desde la última dosis (v. Genotoxicidad y Toxicidad para la función reproductora).
Hombres: Se indicará a los pacientes varones con parejas femeninas que pudieran quedarse embarazadas que deben utilizar un método anticonceptivo eficaz durante el tratamiento con Polivy y hasta que hayan pasado al menos 6 meses desde la última dosis (v. Genotoxicidad y Toxicidad para la función reproductora).
Embarazo: No se recomienda administrar Polivy durante el embarazo, salvo que el beneficio potencial para la madre sea mayor que el riesgo para el feto. Según los resultados de estudios en animales y el mecanismo de acción del fármaco, Polivy puede perjudicar al feto (v. Mecanismo de acción).
Datos de los estudios en animales: En los estudios en animales, la monometilauristatina E (MMAE) provocó manifestaciones de genotoxicidad y toxicidad embriofetal (v. Genotoxicidad y Toxicidad para la función reproductora).
Parto: No se ha determinado que sea seguro utilizar Polivy durante el parto.
Lactancia: No se sabe si el polatuzumab vedotina se excreta en la leche materna humana. No se han llevado a cabo estudios para evaluar la repercusión de Polivy sobre la producción de leche o su presencia en la leche materna. Dado que muchos fármacos se excretan en la leche materna y que cabe la posibilidad de que los bebés amamantados presenten reacciones adversas graves debido a Polivy, las mujeres deben dejar de amamantar mientras estén en tratamiento con Polivy.
Uso en pediatría: No se han determinado la seguridad y la eficacia de Polivy en pacientes pediátricos menores de 18 años.
Uso en geriatría: No se observaron diferencias generales en la seguridad o la eficacia entre los pacientes ≥65 años y los pacientes más jóvenes (v. Pautas posológicas especiales y Farmacocinética en poblaciones especiales).
Disfunción renal: No se han estudiado de manera formal la seguridad y la eficacia de Polivy en pacientes con ClCr <30 ml/min (v. Pautas posológicas especiales y Farmacocinética en poblaciones especiales).
Disfunción hepática: No se han estudiado de manera formal la seguridad y la eficacia de Polivy en pacientes con concentraciones de AST >2,5×LSN, ALT >2,5×LSN o bilirrubina total >1,5×LSN. En estos pacientes se debe vigilar la aparición de reacciones adversas después del tratamiento (véanse los apartados 2.2.1 Pautas posológicas especiales y 3.2.5 Farmacocinética en poblaciones especiales).
DATOS FARMACÉUTICOS: Conservación: Viales: Conserve los viales sin abrir a una temperatura de entre 2 °C y 8 °C.
Mantenga los viales en sus cajas de cartón para resguardarlos de la luz.
No los congele. No los agite.
Periodo de validez: Conforme al registro local.
Este medicamento no debe usarse después de la fecha de caducidad, indicada con «VEN» en el envase.
Periodo de validez del producto reconstituido y la solución para infusión: Véase el apartado Instrucciones especiales de uso, manipulación y eliminación.
La solución reconstituida y la solución para infusión no deben congelarse ni exponerse a la luz solar directa.
PROPIEDADES FARMACOCINÉTICAS: La exposición plasmática a la MMAE conjugada con el anticuerpo (MMAEac) aumentó de manera proporcional a la dosis en el intervalo de dosis de polatuzumab vedotina comprendido entre 0,1 y 2,4 mg/kg. Tras la primera dosis de 1,8 mg/kg de polatuzumab vedotina, la concentración máxima (Cmáx) media de MMAEac era de 803 (± 233) ng/ml y el área bajo la curva de la concentración en función del tiempo desde el tiempo 0 hasta el infinito (ABCinf) era de 1860 (± 966) día*ng/ml. Según el análisis farmacocinético poblacional, en el ciclo 3, el ABC de la MMAEac era un 30% mayor, aproximadamente, que la del ciclo 1 y equivalía a más del 90% del ABC del ciclo 6. En el ciclo 6, la semivida terminal de la MMAEac era de unos 12 días (IC95%: 8,119,5 días).
Las exposiciones a la MMAE no conjugada, que es el componente citotóxico del polatuzumab vedotina, aumentaron de manera proporcional a la dosis en el intervalo de dosis de polatuzumab vedotina comprendido entre 0,1 y 2,4 mg/kg. Las concentraciones plasmáticas de MMAE siguieron una cinética limitada por la velocidad de formación. Tras la primera dosis de 1,8 mg/kg de polatuzumab vedotina, la Cmáx era de 6,82 (± 4,73) ng/ml, el tiempo transcurrido hasta alcanzar la concentración plasmática máxima era de 2,5 días, aproximadamente, y la semivida terminal era de 4 días, aproximadamente. Las exposiciones plasmáticas a la MMAE no conjugada eran <3% de las exposiciones a la MMAEac. Según el análisis farmacocinético poblacional, la exposición plasmática a la MMAE no conjugada (ABC y Cmáx) disminuye tras la administración de varias dosis a intervalos de tres semanas.
Absorción: Polivy se administra como infusión intravenosa. No se han llevado a cabo estudios con otras vías de administración.
Distribución: La estimación poblacional del volumen de distribución central de la MMAEac era de 3,15 l, que se aproxima al volumen plasmático.
In vitro, la MMAE muestra un grado moderado de unión (71%77%) a las proteínas del plasma humano. In vitro, la MMAE no se reparte en grado importante en los eritrocitos humanos; el coeficiente de reparto entre la sangre y el plasma está comprendido entre 1,34 y 1,65.
Los datos obtenidos in vitro indican que la MMAE es un sustrato de la glucoproteína P (P-gp) pero, en concentraciones de trascendencia clínica, no la inhibe.
Metabolismo: Se prevé que, en los pacientes, el polatuzumab vedotina se degrade catabólicamente dando lugar a péptidos pequeños, aminoácidos, MMAE no conjugada y catabolitos relacionados con la MMAE no conjugada.
Según los estudios in vitro, la MMAE es un sustrato de las CYP3A4/5, pero no induce las principales isoenzimas del citocromo P450 (CYP). La MMAE es un inhibidor cronodependiente débil de las CYP3A4/5 pero, en concentraciones de trascendencia clínica, no las inhibe competitivamente.
La MMAE no inhibe la CYP1A2, la CYP2B6, la CYP2C8, la CYP2C9, la CYP2C19 ni la CYP2D6.
Eliminación: Según el análisis farmacocinético poblacional, el conjugado (MMAEac) se elimina fundamentalmente por una vía de aclaramiento lineal inespecífica cuantificada en 0,9 l/día.
En estudios in vivo en los que se administró polatuzumab vedotina (radiomarcado en la MMAE) a ratas, la mayor parte de la radioactividad se excretaba en las heces y en menor proporción en la orina.
Farmacocinética en poblaciones especiales:
Población pediátrica: No se han llevado a cabo estudios para investigar la farmacocinética de Polivy en pacientes pediátricos (<18 años).
Población geriátrica: Según un análisis farmacocinético poblacional llevado a cabo en pacientes de entre 20 y 89 años, la edad no afecta a la farmacocinética de la MMAEac ni la MMAE no conjugada. No se observaron diferencias significativas de la farmacocinética de la MMAEac y la MMAE no conjugada entre los pacientes menores de 65 años (n=187) y los de 65 años en adelante (n=273).
Disfunción renal: Según un análisis farmacocinético poblacional, entre los pacientes con disfunción renal leve (ClCr 60-89 ml/min, n=161) o moderada (ClCr 30-59 ml/min, n=109), las exposiciones a la MMAEac y la MMAE no conjugada son similares a las de los pacientes con función renal normal (ClCr ≥90 ml/min, n=185). No hay datos suficientes para evaluar la repercusión de la disfunción renal grave (ClCr 15-29 ml/min, n=3) sobre la farmacocinética. No se dispone de datos de pacientes con insuficiencia renal terminal o en diálisis (v. Posología y forma de administración).
Disfunción hepática: Según un análisis farmacocinético poblacional, en los pacientes con disfunción hepática leve (AST >1,02,5×LSN o ALT >1,02,5×LSN o bilirrubina total >1,01,5×LSN, n=54), la exposición a la MMAEac era similar a la de los pacientes con función hepática normal (n=399), mientras que el ABC de la MMAE no conjugada era un 40% más alta.
No hay datos suficientes para evaluar la repercusión de la disfunción hepática moderada (bilirrubina total >1,53×LSN, n=2) sobre la farmacocinética. No se dispone de datos de pacientes con disfunción hepática grave o receptores de un trasplante hepático (v. Posología y forma de administración).
CONTRAINDICACIONES: Polivy está contraindicado en los pacientes con hipersensibilidad conocida al polatuzumab vedotina o a cualquiera de los excipientes.
INSTRUCCIONES ESPECIALES DE USO, MANIPULACIÓN Y ELIMINACIÓN: Antes de administrar Polivy, un profesional sanitario debe reconstituirlo con agua estéril para preparaciones inyectables y diluirlo en una bolsa de infusión i.v. que contenga una solución de cloruro de sodio al 0,9%, de cloruro de sodio al 0,45% o de glucosa al 5%.
La reconstitución y dilución de Polivy debe realizarse siguiendo una técnica aséptica. Deben utilizarse los procedimientos adecuados para la preparación de productos antineoplásicos.
El producto reconstituido no contiene conservantes y está destinado únicamente a la administración de una sola dosis. Deseche todo el producto sobrante.
Para administrar Polivy, una vez diluido, debe utilizarse una vía de infusión intravenosa reservada al mismo y equipada con un filtro en línea o adicional (tamaño de poro: 0,2 o 0,22 μm) estéril, apirógeno y con baja afinidad por las proteínas, y un catéter.
Reconstitución: Con una jeringa estéril, inyecte lentamente 7,2 ml de agua estéril para preparaciones inyectables en el vial de 140 mg de Polivy para obtener una solución monodosis que contendrá 20 mg/ml de polatuzumab vedotina. Dirija el chorro de agua hacia la pared del vial, no directamente sobre el polvo compacto liofilizado.
Mueva suavemente el vial en círculos hasta que el polvo compacto se disuelva por completo. No agite el vial.
Inspeccione la solución reconstituida y compruebe que no presenta cambios de color ni contiene partículas. La solución reconstituida debe ser incolora o ligeramente parduzca, y transparente o ligeramente opalescente, y no debe contener partículas visibles. Si la solución reconstituida ha cambiado de color, está turbia o contiene partículas visibles, no la utilice.
Desde el punto de vista microbiológico, es preferible que la solución reconstituida se utilice de inmediato. Si no se utiliza de inmediato, el tiempo y las condiciones de conservación de la solución reconstituida antes de su uso serán responsabilidad del usuario y normalmente no superarán las 24 horas a una temperatura de entre 2 °C y 8 °C, salvo que la reconstitución se haya llevado a cabo en condiciones de asepsia controladas y validadas.
Se ha comprobado que la solución reconstituida es química y físicamente estable durante un periodo de hasta 72 horas a entre 2 °C y 8 °C y durante un periodo de hasta 24 horas a temperatura entre 9 °C y 25 °C.
Dilución:
1. El polatuzumab vedotina debe diluirse hasta una concentración final de entre 0,72 y 2,7 mg/ml en una bolsa de infusión i.v. con un volumen mínimo de 50 ml que contenga una solución de cloruro de sodio al 0,9%, de cloruro de sodio al 0,45% o de glucosa al 5%.
2. Calcule el volumen de solución reconstituida de 20 mg/ml necesario según la dosis que deba administrar:
|
Volumen = |
Dosis de Polivy (1,8 o 1,4 mg/kg) × peso del paciente (kg) |
|
Concentración del vial reconstituido (20 mg/ml) |
3. Con una jeringa estéril, extraiga y deseche de la bolsa de infusión i.v. un volumen de diluyente equivalente al volumen necesario de solución reconstituida.
4. Con una jeringa estéril, extraiga del vial de Polivy el volumen necesario de solución reconstituida y dilúyalo en el contenido de la bolsa de infusión i.v. Deseche todo el producto que quede en el vial.
5. Mezcle suavemente el contenido de la bolsa de infusión i.v. invirtiendo la bolsa lentamente. No agite la bolsa.
6. Inspeccione la bolsa de infusión i.v.; si la solución contiene partículas, deséchela.
Desde el punto de vista microbiológico, es preferible que la solución para infusión preparada se utilice de inmediato. Si no se utiliza de inmediato, el tiempo y las condiciones de conservación de la solución para infusión antes de su uso serán responsabilidad del usuario y normalmente no superarán las 24 horas a una temperatura de entre 2 °C y 8 °C, salvo que la dilución se haya llevado a cabo en condiciones de asepsia controladas y validadas. Se ha comprobado que, una vez preparada, la solución para infusión mantiene una estabilidad química y física aceptable durante los tiempos indicados en la tabla 5. Si el tiempo de conservación supera los indicados, deseche la solución para infusión. No congele la solución para infusión ni la exponga a la luz solar directa.
|
Tabla 5. Tiempos comprobados durante los que la solución para infusión preparada mantiene una estabilidad química y física aceptable |
|
|
Diluyente utilizado para preparar la solución para infusión |
Condiciones de conservación de la solución para infusión1 |
|
Cloruro de sodio al 0,9% |
Hasta 24 horas a entre 2 °C y 8 °C o hasta 4 horas a temperatura entre 9 °C y 25 °C |
|
Cloruro de sodio al 0,45% |
Hasta 72 horas a entre 2 °C y 8 °C o hasta 8 horas a temperatura entre 9 °C y 25 °C |
|
Glucosa al 5% |
Hasta 72 horas a entre 2 °C y 8 °C o hasta 8 horas a temperatura entre 9 °C y 25 °C |
|
1Para garantizar la estabilidad del producto, no supere los tiempos de conservación especificados. |
|
Evite transportar la solución para infusión preparada, ya que si se agita pueden formarse agregados. Si va a transportarla, extraiga el aire de la bolsa de infusión y limite la duración del transporte a 30 minutos a una temperatura comprendida entre 9 °C y 25 °C o a 2 horas a una temperatura comprendida entre 2 °C y 8 °C. Si se ha extraído el aire, hará falta un equipo de infusión provisto de un punzón perforador con toma de aire para garantizar la administración de la dosis correcta durante la infusión.
Incompatibilidades: No mezcle Polivy con otros medicamentos ni lo administre a través de la misma vía de infusión.
No se han observado incompatibilidades entre Polivy y las bolsas de infusión i.v. cuyo material de contacto con el producto es poli(cloruro de vinilo) (PVC) o una poliolefina (PO) como el polietileno (PE) o el polipropileno (PP). Tampoco se han observado incompatibilidades con equipos u otros accesorios para infusión i.v. cuyo material de contacto con el producto es PVC, PE, poliuretano (PU), polibutadieno (PBD), acrilonitrilo butadieno estireno (ABS), policarbonato (PC), polieteruretano (PEU) o etileno-propileno fluorado (FEP) y provistos de filtros de membrana de polietersulfona (PES) o polisulfona (PSU).
Eliminación del medicamento sobrante o caducado: La emisión de medicamentos al medio ambiente debe reducirse al mínimo. Evítese tirarlos por los desagües o a la basura doméstica.
Las jeringas y todo el material médico punzocortante deben utilizarse y eliminarse siguiendo al pie de la letra las instrucciones siguientes:
Las agujas y las jeringas nunca deben reutilizarse. Coloque todas las agujas y jeringas usadas en un recipiente especial para material punzocortante (imperforable).
El medicamento que no se haya utilizado y el material de desecho deben eliminarse de acuerdo con las normas locales.
CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: Polivy puede afectar en grado leve a la capacidad para conducir y utilizar máquinas.
Durante el tratamiento con Polivy pueden presentarse reacciones relacionadas con la infusión, neuropatía periférica, fatiga y mareo (v. Advertencias y precauciones y Reacciones adversas).
PROPIEDADES Y EFECTOS FARMACOLÓGICOS:
Propiedades farmacodinámicas:
Mecanismo de acción: El polatuzumab vedotina es un conjugado de anticuerpo y fármaco dirigido contra CD79b que libera un potente antimitótico (la monometilauristatina E, o MMAE) preferentemente en los linfocitos B, lo cual causa la muerte de los linfocitos B malignos. La molécula de polatuzumab vedotina consiste en MMAE unida covalentemente a un anticuerpo monoclonal humanizado de tipo inmunoglobulina G1 (IgG1) por medio de un conector escindible. El anticuerpo monoclonal se une con gran afinidad y selectividad a CD79b, que es un componente del receptor de antígeno de los linfocitos B situado en la superficie celular. La expresión de CD79b se limita a las células normales de la estirpe de los linfocitos B (con la excepción de los plasmocitos) y a los linfocitos B malignos; se expresa en más del 95% de los LDLBG. Cuando el polatuzumab vedotina se une a CD79b, se interioriza rápidamente y las proteasas lisosómicas escinden el conector para permitir la liberación intracelular de la MMAE. Esta se une a los microtúbulos y causa la muerte de las células que estén en división al inhibir la división e inducir la apoptosis.
Ensayos clínicos/Estudios de eficacia: Se evaluó la eficacia de Polivy en un ensayo internacional, multicéntrico y sin enmascaramiento (GO29365) que comprendía una cohorte aleatorizada de 80 pacientes con LDLBG que habían sido tratados anteriormente. Se asignó aleatoriamente a los pacientes, en proporción 1:1, a recibir Polivy más BR o bien solo BR durante seis ciclos de 21 días. Se estratificó a los pacientes según la duración de la respuesta al último de los tratamientos previos: ≤12 meses o >12 meses.
Los pacientes aptos para participar en el estudio no eran candidatos a recibir un autotrasplante de precursores hematopoyéticos y presentaban un LDLBG recidivante o resistente al tratamiento tras haber recibido anteriormente al menos un esquema quimioterápico por vía sistémica. Se excluyó del estudio a los pacientes que habían recibido anteriormente un alotrasplante de precursores hematopoyéticos o presentaban un linfoma del sistema nervioso central, un linfoma folicular (LF) transformado o un LF de grado 3b.
Polivy se administró en dosis de 1,8 mg/kg por vía intravenosa el día 2 del ciclo 1 y el día 1 de los ciclos 2 a 6. La bendamustina se administró en dosis diarias de 90 mg/m2 por vía intravenosa los días 2 y 3 del ciclo 1 y los días 1 y 2 de los ciclos 2 a 6. El rituximab se administró en dosis de 375 mg/m2 por vía intravenosa el día 1 de los ciclos 1 a 6.
Los dos grupos de tratamiento estaban en general equilibrados en cuanto a las características demográficas y nosológicas al inicio. La mediana de la edad era de 69 años (intervalo: entre 30 y 86 años), el 71% de los pacientes eran de raza blanca y el 66% eran varones. La mayoría de los pacientes (98%) presentaban LDLBG sin especificar. En conjunto, el 48% de los pacientes presentaban un LDLBG con linfocitos similares a los linfocitos B activados (LBA) y el 40% presentaban un LDLBG con linfocitos similares a los de los centros germinales. Las razones principales por las que los pacientes no eran candidatos a recibir un trasplante de precursores hematopoyéticos eran la edad (40%), la respuesta insuficiente al tratamiento de rescate (26%) y el fracaso de un trasplante anterior (20%). La mediana del número de tratamientos anteriores era de 2 (intervalo: 1-7): el 29% (n=23) de los pacientes habían recibido un tratamiento anterior, el 25% (n=20) habían recibido 2 y el 46% (n=37) habían recibido 3 o más. El 80% de los pacientes presentaban un LDLBG resistente al tratamiento.
La variable principal de valoración del estudio era la tasa de respuesta completa (RC) al final del tratamiento (entre 6 y 8 semanas después del día 1 del ciclo 6 o del último tratamiento del estudio), evaluada por un comité independiente de evaluación (CIE). Los resultados de la eficacia se resumen en la tabla 4 y en las figuras 1 a 3.
|
Tabla 4. Resumen de la eficacia en los pacientes con LDLBG tratado anteriormente (estudio GO29365) |
||
|
Polivy + bendamustina + rituximab N= 40 |
Bendamustina + rituximab N= 40 |
|
|
Mediana del tiempo de observación: 22 meses |
||
|
Variable principal de valoración |
||
|
Tasa de respuesta completa* (evaluada por el CIE) al final del tratamiento** |
||
|
Pacientes con respuesta al tratamiento (%) |
16 (40,0) |
7 (17,5) |
|
Diferencia en la tasa de respuesta (%) [IC95%] |
22,5 [2,6, 40,2] |
|
|
p (prueba de ?2 de CMH***) |
0,0261 |
|
|
Variables clave de valoración |
||
|
Supervivencia global (SVG) |
||
|
Número (%) de pacientes que presentaron el evento |
23 (57,5) |
28 (70,0) |
|
Mediana de la SVG (IC95%), meses HR [IC95%] |
12,4 (9,0, NE) |
4,7 (3,7, 8,3) |
|
0,42 [0,24, 0,75] |
||
|
p (prueba del orden logarítmico estratificada***) |
0,0023¶ |
|
|
Supervivencia sin progresión (SVSP) (evaluada por el investigador) |
||
|
Número (%) de pacientes que presentaron el evento |
27 (67,5) |
35 (87,5) |
|
Mediana de la SVSP (IC95%), meses |
7,6 (6,0, 17,0) |
2,0 (1,5, 3,7) |
|
HR [IC95%] |
0,34 [0,20, 0,57] |
|
|
p (prueba del orden logarítmico estratificada***) |
<0,0001 |
|
|
Duración de la respuesta (DR) (evaluada por el investigador) |
||
|
Número de pacientes incluidos en el análisis Número (%) de pacientes que presentaron el evento |
28 17 (60,7) |
13 11 (84,6) |
|
Mediana de la DR (IC95%), meses HR [IC95%] |
10,3 (5,6, NE) |
4,1 (2,6, 12,7) |
|
0,44 [0,20, 0,95] |
||
|
p (prueba del orden logarítmico estratificada***) |
0,0321 |
|
|
Tasa de respuesta global* (evaluada por el investigador) al final del tratamiento** |
||
|
Pacientes con respuesta al tratamiento (%) (RC, RP) |
19 (47,5) |
7 (17,5) |
|
Diferencia en la tasa de respuesta (%) [IC95%] |
30,0 [9,5, 47,4] |
|
|
p (prueba de ?2 de CMH***) |
0,0036 |
|
|
Respuesta completa (%) (RC) |
17 (42,5) |
6 (15,0) |
|
Diferencia en la tasa de respuesta (%) [IC95%] |
27,5 [7,7, 44,7] |
|
|
p (prueba de ?2 de CMH***) |
0,0061 |
|
|
Respuesta parcial (%) (RP) IC95% (Clopper-Pearson) |
2 (5,0) [0,6, 16,9] |
1 (2,5) [0,06, 13,2] |
|
Mejor tasa de respuesta global* (evaluada por el investigador) |
||
|
Pacientes con respuesta al tratamiento (%) (RC, RP) |
28 (70,0) |
13 (32,5) |
|
Diferencia en la tasa de respuesta (%) [IC95%] |
37,5 [15,6, 54,7] |
|
|
Respuesta completa (%) (RC) |
23 (57,5) |
8 (20,0) |
|
IC95% (Clopper-Pearson) |
[40,9, 73,0] |
[9,1, 35,7] |
|
Respuesta parcial (%) (RP) IC95% (Clopper-Pearson) |
5 (12,5) [4,2, 26,8] |
5 (12,5%) [4,2, 26,8] |
|
CIE: comité independiente de evaluación; CMH: Cochran-Mantel-Haenszel; DR: duración de la respuesta; HR: cociente de riesgos instantáneos (en inglés: hazard ratio); IC: intervalo de confianza; NE: no evaluable; SVG: supervivencia global; SVSP: supervivencia sin progresión. *Según los criterios modificados de Lugano (2014): Se exigía confirmación en la médula ósea de la RC basada en la tomografía por emisión de positrones-tomografía computarizada (PET-TAC). La RP basada en la PET-TAC debía cumplir tanto los criterios de la PET-TAC como los de la TAC [56]. **Entre 6 y 8 semanas después del día 1 del ciclo 6 o del último tratamiento del estudio. ***Estratificación según la duración de la respuesta al tratamiento previo (≤12 meses o >12 meses). |
||
Figura 1. Curvas de Kaplan-Meier de la supervivencia global
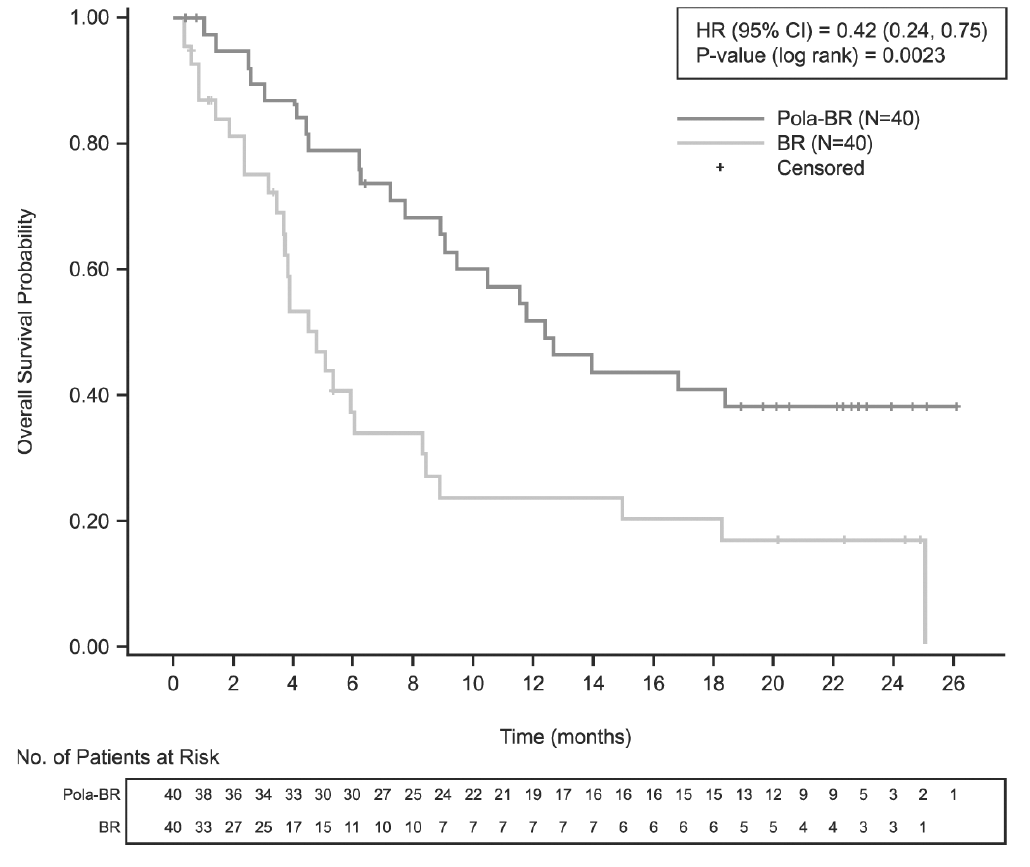
Eje de ordenadas: Overall Survival Probability = Probabilidad de supervivencia global. Eje de abscisas: Time (months) = Tiempo (meses). Leyenda: HR (95%CI) = HR (IC95%); P-value (log-rank) = p (prueba del orden logarítmico); Censored = Datos censurados para el análisis. Pie de la tabla: No. of Patients at Risk = Núm. de pacientes en riesgo. BR: bendamustina y rituximab; HR: cociente de riesgos instantáneos; Núm.: número; Pola: Polivy.
Figura 2. Curvas de Kaplan-Meier de la supervivencia sin progresión evaluad por el investigador
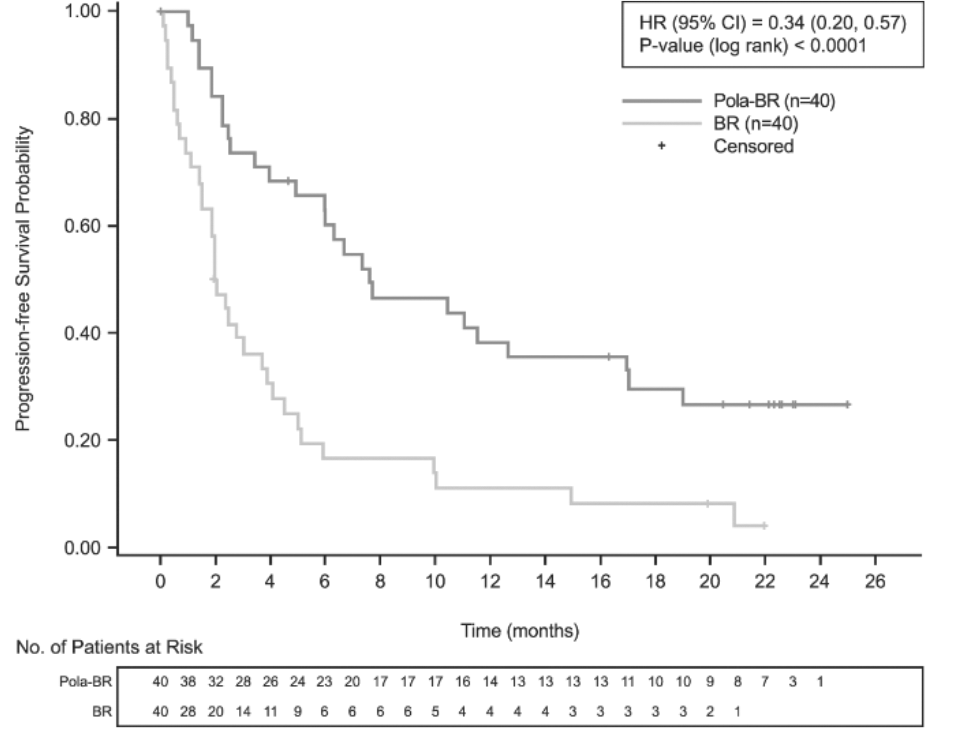
Eje de ordenadas: Progression-free Survival Probability = Probabilidad de supervivencia sin progresión. Eje de abscisas: Time (months) = Tiempo (meses). Leyenda: HR (95%CI) = HR (IC95%); P-value (log-rank) = p (prueba del orden logarítmico); Censored = Datos censurados para el análisis. Pie de la tabla: No. of Patients at Risk = Núm. de pacientes en riesgo.
BR: bendamustina y rituximab; HR: cociente de riesgos instantáneos; Núm.: número; Pola: Polivy.
Resultados de los análisis por subgrupos: Los resultados de los análisis por subgrupos de la supervivencia global concordaban con los observados en la población global con LDLBG (véase la figura 3 a continuación).
Figura 3. Gráfico de bosque de la supervivencia global
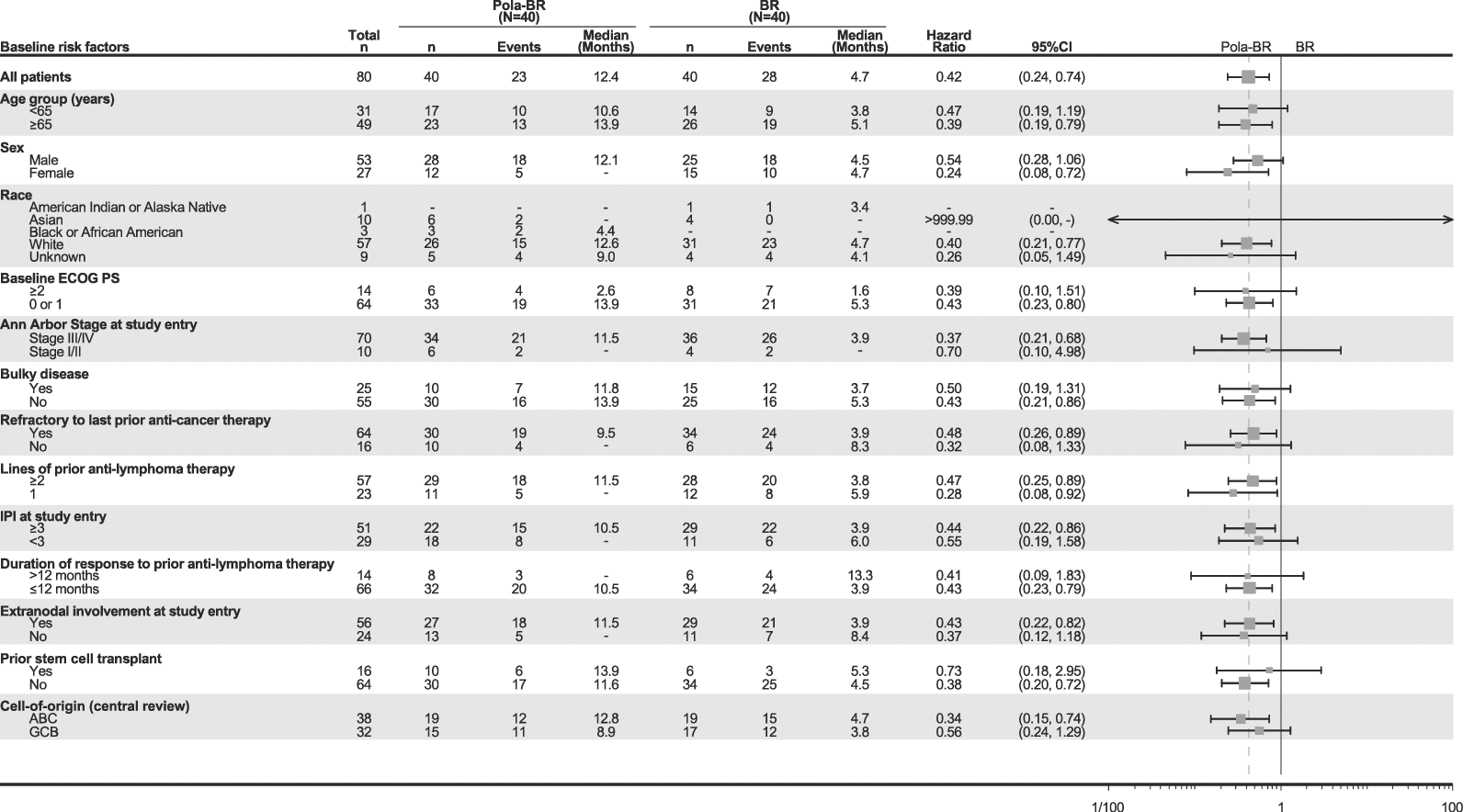
Primera columna: Baseline risk factors = Factores de riesgo iniciales; All patients = Todos los pacientes; Age group (years) = Grupo etario (años); Sex = Sexo; Male = Hombre; Female = Mujer; Race = Raza; American Indian or Alaska Native = Indios estadounidenses o nativos de Alaska; Asian = Asiáticos; Black or African American = Negros o afroestadounidenses; White = Blancos; Unknown = Desconocida; Baseline ECOG PS = Puntuación inicial de la EF ECOG; Ann Arbor Stage at study entry = Estadio de Ann Arbor al ingresar en el estudio; Stage = Estadio; Bulky disease = Gran masa tumoral; Yes = Sí; Refractory to last prior anti-cancer therapy = Resistente al último tratamiento antineoplásico previo; Lines of prior anti-lymphoma therapy = Líneas de tratamiento antilinfomatoso anteriores; IPI at study entry = IPI al ingresar en el estudio; Duration of response to prior anti-lymphoma therapy = Duración de la respuesta al tratamiento antilinfomatoso anterior; Extranodal involvement at study entry = Afectación extraganglionar al ingresar en el estudio; Prior stem cell transplant = Antecedentes de trasplante de precursores hematopoyéticos; Cell-of-origin (central review) = Célula de origen (examen centralizado); ABC = LBA; GCB = LBCG. Encabezamientos de las columnas: Events = Eventos; Median (months) = Mediana (meses); Hazard ratio = Cociente de riesgos instantáneos; 95%CI = IC95%.
BR: bendamustina y rituximab; EF ECOG: escala funcional del Eastern Cooperative Oncology Group (de los EE.UU.); IC: intervalo de confianza; IPI: índice pronóstico internacional; LBA: linfocitos B activados; LBCG: linfocitos B similares a los de los centros germinales; Pola: Polivy.
Inmunogenia: Como ocurre con todas las proteínas terapéuticas, los pacientes tratados con polatuzumab vedotina podrían presentar una respuesta inmunitaria. Considerando todos los grupos del estudio GO29365, 8 de 134 (6,0%) pacientes tuvieron un resultado positivo en al menos una de las pruebas de detección de anticuerpos contra el polatuzumab vedotina realizadas después del inicio. En el conjunto de los siete ensayos clínicos, 14 de 536 (2,6%) pacientes tuvieron un resultado positivo en al menos una de las pruebas de detección de anticuerpos contra el polatuzumab vedotina realizadas después del inicio. Dado el reducido número de pacientes que presentaron anticuerpos contra el polatuzumab vedotina, no es posible extraer conclusiones relativas a un posible efecto de la inmunogenia sobre la eficacia o la seguridad.
Los resultados de las pruebas de inmunogenia dependen en gran medida de varios factores, como la sensibilidad y la especificidad de la prueba, el método seguido para realizarla, la manipulación de las muestras, el cronograma de obtención de estas, la coadministración de otros medicamentos y la enfermedad de base. Por ello, comparar la incidencia de anticuerpos contra el polatuzumab vedotina con la incidencia de anticuerpos contra otros productos puede inducir a error.
REACCIONES ADVERSAS: Ensayos clínicos: Resumen del perfil de seguridad: Se calcula que 588 pacientes han recibido Polivy durante el programa de desarrollo clínico de este medicamento, considerado en su conjunto. Las reacciones adversas (RA) descritas en este apartado se identificaron durante el tratamiento y el seguimiento de los pacientes con linfoma difuso de linfocitos B grandes (LDLBG) que habían sido tratados anteriormente y que participaron en el ensayo clínico fundamental GO29365. Están incluidos los pacientes de la fase de preinclusión para el estudio de la seguridad (n=6) y los pacientes aleatorizados (n=39) que recibieron Polivy en combinación con bendamustina y rituximab (BR), en comparación con los pacientes aleatorizados (n=39) que recibieron solo BR [28]. Los pacientes asignados aleatoriamente al grupo del tratamiento con Polivy recibieron una cifra mediana de 5 ciclos de tratamiento, mientras que los asignados aleatoriamente al grupo del tratamiento comparativo recibieron una cifra mediana de 3 ciclos de tratamiento.
Resumen tabular de las RA registradas en los ensayos clínicos: En la tabla 3 se enumeran las RA según las categorías del MedDRA de órgano, aparato o sistema afectado (en inglés: system organ class, SOC).
Las RA notificadas con mayor frecuencia (≥30%) en los pacientes tratados con Polivy en combinación con BR eran anemia, trombocitopenia, neutropenia, fatiga, diarrea, náuseas y pirexia (fiebre) [28]. Se notificaron eventos adversos graves en el 64,4% de los pacientes tratados con Polivy más BR, que consistieron en neutropenia febril (11,1%), pirexia (8,9%), neumonía (8,9%), anemia (4,4%), hemorragia de úlcera de duodeno (4,4%), sepsis (4,4%) y trombocitopenia (4,4%).
Las RA que obligaron a suspender definitivamente el tratamiento en >5% de los pacientes fueron la trombocitopenia (8,9%) y la neutropenia (6,7%).
|
Tabla 3. Resumen de las reacciones adversas aparecidas en pacientes con LDLBG que habían sido tratados anteriormente y estaban recibiendo Polivy en combinación con BR |
||||
|
Reacciones adversas |
Polivy + bendamustina + rituximab N=45 |
Bendamustina + rituximab N=39 |
||
|
SOC |
Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
Infecciones e infestaciones |
||||
|
Neumoníaa |
15,6 |
6,7 |
10,3 |
0 |
|
Trastornos de la sangre y el sistema linfático |
||||
|
Anemia |
46,7 |
24,4 |
25,6 |
17,9 |
|
Neutropenia |
46,7 |
40,0 |
38,5 |
33,3 |
|
Trombocitopenia |
46,7 |
37,8 |
28,2 |
23,1 |
|
Neutropenia febril |
11,1 |
11,1 |
12,8 |
12,8 |
|
Leucopenia |
11,1 |
6,7 |
12,8 |
7,7 |
|
Linfopenia |
11,1 |
11,1 |
0 |
0 |
|
Trastornos del metabolismo y la nutrición |
||||
|
Apetito disminuido |
26,7 |
2,2 |
20,5 |
0 |
|
Hipopotasemia |
15,6 |
6,7 |
7,7 |
2,6 |
|
Hipoalbuminemia |
13,3 |
2,2 |
5,1 |
0 |
|
Hipocalcemia |
11,1 |
2,2 |
2,6 |
0 |
|
Trastornos del sistema nervioso |
||||
|
Neuropatía periférica |
20,0 |
0 |
2,6 |
0 |
|
Mareo |
13,3 |
0 |
7,7 |
0 |
|
Neuropatía periférica sensitiva |
13,3 |
0 |
0 |
0 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Tos |
15,6 |
0 |
20,5 |
0 |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
37,8 |
4,4 |
28,2 |
5,1 |
|
Náuseas |
33,3 |
0 |
41,0 |
0 |
|
Estreñimiento |
17,8 |
0 |
20,5 |
2,6 |
|
Vómitos |
17,8 |
2,2 |
12,8 |
0 |
|
Dolor abdominal |
11,1 |
4,4 |
10,3 |
2,6 |
|
Dolor en la zona superior del abdomen |
11,1 |
2,2 |
5,1 |
0 |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Prurito |
13,3 |
0 |
10,3 |
2,6 |
|
Trastornos generales y alteraciones en el lugar de administración |
||||
|
Fatiga |
40,0 |
4,4 |
35,9 |
2,6 |
|
Pirexia |
33,3 |
2,2 |
23,1 |
0 |
|
Astenia |
11,1 |
0 |
15,4 |
0 |
|
Escalofríos |
11,1 |
0 |
7,7 |
0 |
|
Exploraciones complementarias |
||||
|
Peso disminuido |
15,6 |
2,2 |
7,7 |
2,6 |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
||||
|
Reacción relacionada con la infusiónb |
33,3 |
6,7 |
23,1 |
10,3 |
|
a RA asociada a desenlace mortal. b Definida como todos los eventos adversos notificados como relacionados con el tratamiento del estudio en las 24 horas posteriores a la infusión de este. |
||||
Descripción de algunas reacciones adversas registradas en los ensayos clínicos: Mielodepresión: La neutropenia obligó a suspender definitivamente la administración de Polivy en el 8,9% de los pacientes del grupo de Polivy más BR, frente al 2,6% de los pacientes del grupo de BR que tuvieron que suspender definitivamente el tratamiento. Los eventos de trombocitopenia obligaron a suspender definitivamente el tratamiento en el 11,1% de los pacientes del grupo de Polivy más BR y el 5,1% de los pacientes del grupo de BR. La anemia no obligó a suspender definitivamente el tratamiento en ningún paciente, ni del grupo de Polivy más BR ni del grupo de BR.
Neuropatía periférica (NP): En el grupo de Polivy más BR, se notificaron eventos de NP de grado 1 en el 26,7% de los pacientes y de grado 2 en el 13,3%. En el grupo de BR se notificaron eventos de NP de grado 1 en el 2,6% de los pacientes y de grado 2 en el 5,1%. Ni en el grupo de Polivy más BR ni en el de BR se notificaron eventos de NP de grado 3-5. La NP obligó a suspender definitivamente el tratamiento con Polivy en el 2,2% de los pacientes y a reducir la dosis en el 4,4%. En ningún paciente del grupo de BR se suspendió definitivamente el tratamiento o se redujo la dosis debido a NP. En el grupo de Polivy más BR, la mediana del tiempo transcurrido entre el inicio y el primer evento de NP fue de 1,8 meses, y en el 61,1% de los pacientes con eventos de NP se notificó la resolución del evento (v. Advertencias y precauciones).
Infecciones: Se notificaron infecciones, que incluyen la neumonía y otros tipos de infecciones, en el 53,3% de los pacientes del grupo de Polivy más BR y el 51,3% de los pacientes del grupo de BR. En el grupo de Polivy más BR, se notificaron infecciones graves en el 28,9% de los pacientes e infecciones mortales en el 8,9%. En el grupo de BR, se notificaron infecciones graves en el 30,8% de los pacientes e infecciones mortales en el 10,3%. La infección obligó a suspender definitivamente el tratamiento en un paciente (2,2%) del grupo de Polivy más BR, frente al 5,1% de los pacientes en el grupo de BR (v. Advertencias y precauciones).
Leucoencefalopatía multifocal progresiva (LMP): Se produjo un caso de LMP, que resultó mortal, en un paciente tratado con Polivy más bendamustina y obinutuzumab. Este paciente había recibido anteriormente tres líneas de tratamiento que incluyeron anticuerpos anti-CD20 (v. Advertencias y precauciones).
Hepatotoxicidad: En otro estudio se notificaron dos casos de hepatotoxicidad grave (lesión hepatocelular y esteatosis hepática), ambos reversibles (v. Advertencias y precauciones).
Toxicidad gastrointestinal: Se notificaron eventos de toxicidad gastrointestinal en el 80,0% de los pacientes del grupo de Polivy más BR, frente al 64,1% de los pacientes del grupo de BR. La mayor parte fueron de grado 1-2, y se notificaron eventos de grado 3-4 en el 22,2% de los pacientes del grupo de Polivy más BR, frente al 12,8% de los pacientes del grupo de BR. Los eventos de toxicidad gastrointestinal más frecuentes fueron diarrea y náuseas.
Experiencia poscomercialización: No procede.
INTERACCIONES CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN: No se han llevado a cabo estudios clínicos específicos de las interacciones farmacológicas con la administración de Polivy a seres humanos.
Interacciones farmacológicas con fármacos coadministrados que son inhibidores, inductores o sustratos de las CYP3A: Según las simulaciones del modelo farmacocinético fisiológico de la MMAE liberada a partir del polatuzumab vedotina, los inhibidores potentes de las CYP3A (como el ketoconazol) pueden aumentar en un 48% el área bajo la curva de la concentración en función del tiempo (ABC) de la MMAE no conjugada. Se debe vigilar más estrechamente a los pacientes que estén recibiendo también un inhibidor potente de las CYP3A por si aparecieran señales de toxicidad. Los inductores potentes de las CYP3A (como la rifampicina) pueden disminuir en un 49% el ABC de la MMAE no conjugada.
El modelo no predice que la MMAE no conjugada modifique el ABC de los fármacos coadministrados que sean sustratos de las CYP3A (como el midazolam).
Interacciones farmacológicas del rituximab y la bendamustina en combinación con el polatuzumab vedotina: La coadministración de Polivy no afecta a la farmacocinética del rituximab ni a la de la bendamustina. Según el análisis farmacocinético poblacional, la coadministración de rituximab se asocia a un aumento del área bajo la curva de la concentración plasmática en función del tiempo de la MMAE conjugada con el anticuerpo (MMAEac) del 24% y a una disminución del área bajo la curva de la concentración plasmática en función del tiempo de la MMAE no conjugada del 37%. No es preciso ajustar la dosis.
La bendamustina no afecta al área bajo la curva de la concentración plasmática en función del tiempo de la MMAEac ni de la MMAE no conjugada.
DATOS PRECLÍNICOS SOBRE SEGURIDAD: Carcinogenia: No se han llevado a cabo estudios específicos de carcinogenia en animales con Polivy, MMAE o ambos.
Genotoxicidad: No se han llevado a cabo estudios específicos de mutagenia en animales con Polivy. En la prueba de los micronúcleos llevada a cabo en médula ósea de ratas, la MMAE era genotóxica a través de un mecanismo aneuploidogénico. Este mecanismo concuerda con el efecto farmacológico de la MMAE, que es un fármaco desestabilizador de los microtúbulos. La MMAE no era mutágena en la prueba de retromutaciones bacterianas (prueba de Ames) ni en la prueba de mutaciones directas en células de linfoma murino L5178Y.
Trastornos de la fecundidad: No se han llevado a cabo estudios específicos en animales de los efectos de Polivy sobre la fecundidad. Sin embargo, los estudios de toxicidad con dosis múltiples realizados en ratas indican que el polatuzumab vedotina podría afectar a la función reproductora y la fecundidad masculinas. En el estudio de toxicidad con dosis múltiples de 4 semanas de duración llevado a cabo en ratas, y en el que se administraron semanalmente 2, 6 y 10 mg/kg, se observó una degeneración de los túbulos seminíferos testiculares dependiente de la dosis, con presencia de un contenido luminal anormal en el epidídimo. Las anomalías observadas en los testículos y el epidídimo no remitieron y se correlacionaron con un menor peso testicular y con observaciones macroscópicas de testículos pequeños o blandos en la necropsia de recuperación en machos que recibieron dosis ≥2 mg/kg.
Toxicidad para la función reproductora: No se han llevado a cabo estudios específicos de teratogenia en animales con Polivy. Sin embargo, se evaluó la MMAE en ratas en un estudio del desarrollo embriofetal y la toxicocinética llevado a cabo según las prácticas correctas de laboratorio (en inglés: good laboratory practices, GLP), en el cual ratas gestantes recibieron 2 dosis de 0,2 mg/kg de MMAE por vía intravenosa durante el periodo de organogénesis, los días 6 y 13 de la gestación. La administración de MMAE en dosis de 0,2 mg/kg causó malformaciones fetales externas como lengua protruyente, malrotaciones de los miembros, gastrosquisis y agnatia. La exposición sistémica (ABC) obtenida en ratas con una dosis de MMAE de 0,2 mg/kg equivale aproximadamente al 50% del ABC registrada en los pacientes que recibieron la dosis recomendada de Polivy de 1,8 mg/kg cada 21 días.
ADVERTENCIAS Y PRECAUCIONES: Advertencias y precauciones generales: Mielodepresión: Se han notificado eventos graves e intensos de neutropenia y neutropenia febril en pacientes tratados con Polivy ya desde el primer ciclo de tratamiento (véase el apartado 2.6 Reacciones adversas). Se debe considerar la pertinencia de administrar profilaxis con factor estimulante de las colonias de granulocitos (G-CSF). También puede aparecer trombocitopenia o anemia de grado 3 o 4 con el tratamiento con Polivy (véase el apartado 2.6 Reacciones adversas). Antes de administrar cada dosis de Polivy debe comprobarse el hemograma completo. En los pacientes con neutropenia y trombocitopenia de grado 3 o 4 se debe considerar la pertinencia de hacer controles analíticos más frecuentes y/o de retrasar o suspender definitivamente el tratamiento con Polivy (v. Posología y forma de administración).
Neuropatía periférica: Se ha notificado la aparición de neuropatía periférica en pacientes tratados con Polivy ya desde el primer ciclo de tratamiento, y el riesgo aumenta con las dosis sucesivas (véase el apartado 2.6 Reacciones adversas). En los pacientes que ya padecían anteriormente una neuropatía periférica, esta puede empeorar. La neuropatía periférica notificada en los pacientes tratados con Polivy es predominantemente sensitiva; sin embargo, también se han notificado casos de neuropatía periférica motora y sensitivomotora. Se debe vigilar la aparición de síntomas de neuropatía periférica, como hipoestesia, hiperestesia, parestesia, disestesia, dolor neuropático, sensación de ardor, debilidad o alteración de la marcha. En los pacientes en los que aparezca una neuropatía periférica o que presenten un empeoramiento de la que ya padecían puede ser necesario retrasar el tratamiento con Polivy, reducir la dosis o suspenderlo definitivamente (v. Posología y modo de administración).
Infecciones: Se ha notificado en pacientes tratados con Polivy la aparición de infecciones graves, potencialmente mortales o mortales, incluidas infecciones oportunistas, como neumonía (incluidas la debida a Pneumocystis jirovecii y otras neumonías micóticas), bacteriemia, sepsis, infección herpética e infección por citomegalovirus (v. Reacciones adversas). Durante el tratamiento se debe vigilar estrechamente la aparición de signos de infección bacteriana, micótica o vírica. Debe considerarse la pertinencia de administrar una profilaxis antiinfecciosa. En los pacientes que presenten una infección grave se suspenderá definitivamente la administración de Polivy y de toda quimioterapia concurrente.
Leucoencefalopatía multifocal progresiva (LMP): Se han notificado casos de LMP con el tratamiento con Polivy (v. Reacciones adversas). Se debe vigilar estrechamente la aparición o el empeoramiento de alteraciones neurológicas, cognitivas o conductuales que sean indicativas de LMP. Si se sospecha una LMP, se debe interrumpir la administración de Polivy y de toda quimioterapia concurrente, y en caso de confirmarse el diagnóstico, se suspenderá el tratamiento definitivamente.
Síndrome de lisis tumoral: Los pacientes con gran masa tumoral y un tumor de proliferación rápida pueden tener más riesgo de presentar un síndrome de lisis tumoral. Antes de iniciar el tratamiento con Polivy deben tomarse las medidas adecuadas, de conformidad con las directrices locales. Se debe vigilar estrechamente la aparición de un síndrome de lisis tumoral durante el tratamiento con Polivy.
Toxicidad embriofetal: Teniendo en cuenta el mecanismo de acción y los resultados de los estudios preclínicos, Polivy puede perjudicar al feto si se administra a la embarazada (v. Embarazo, Mecanismo de acción y Toxicidad para la función reproductora). Se debe informar a las embarazadas del riesgo para el feto.
Se indicará a las mujeres con posibilidad de quedar embarazadas que deben utilizar un método anticonceptivo eficaz durante el tratamiento con Polivy y hasta que hayan pasado al menos 9 meses desde la última dosis. Se indicará a los pacientes varones con parejas femeninas que pudieran quedarse embarazadas que deben utilizar un método anticonceptivo eficaz durante el tratamiento con Polivy y hasta que hayan pasado al menos 6 meses desde la última dosis (v. Mujeres y hombres con posibilidad de procrear, Genotoxicidad y Toxicidad para la función reproductora).
Hepatotoxicidad: Se han observado casos graves de hepatotoxicidad compatibles con una lesión hepatocelular, como elevaciones de la concentración de transaminasas o de bilirrubina, en pacientes tratados con Polivy. El riesgo puede ser mayor en los pacientes con una hepatopatía previa o altas concentraciones iniciales de enzimas hepáticas, o que estén recibiendo también otros medicamentos. Se deben vigilar las concentraciones de enzimas hepáticas y de bilirrubina.
Abuso y dependencia del fármaco: Polivy no tiene potencial adictivo ni genera dependencia.
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN: Instrucciones generales: La sustitución por cualquier otro medicamento biológico deberá ser autorizada por el médico prescriptor.
Para evitar los errores de medicación es importante examinar las etiquetas del vial y comprobar que el medicamento que se va a preparar y administrar es Polivy.
El tratamiento con Polivy debe administrarse únicamente bajo la supervisión de un profesional sanitario con experiencia en el tratamiento de pacientes oncológicos.
Polivy debe reconstituirse y diluirse siguiendo una técnica aséptica y bajo la supervisión de un profesional sanitario. Polivy debe administrarse en infusión intravenosa a través de una vía de infusión reservada al mismo y equipada con un filtro en línea o adicional estéril, apirógeno y con baja afinidad por las proteínas (tamaño de poro: 0,2 o 0,22 μm) y un catéter (v. Instrucciones especiales de uso, manipulación y eliminación). No debe administrarse en inyección i.v. lenta ni rápida (en embolada).
La información sobre el rituximab y la bendamustina debe consultarse en las respectivas fichas técnicas En la tabla 2 se especifican las modificaciones de la dosis recomendadas en los pacientes que presenten neutropenia o trombocitopenia.
Dosis recomendada: La dosis recomendada de Polivy es de 1,8 mg/kg administrados en infusión intravenosa cada 21 días, en combinación con bendamustina y rituximab, durante 6 ciclos. El Polivy, la bendamustina y el rituximab se pueden administrar en el orden que se desee el día 1 de cada ciclo. La dosis recomendada de bendamustina es de 90 mg/m2/día el día 1 y el 2 cuando se administra con Polivy y rituximab.
Si el paciente no ha recibido premedicación, antes de comenzar la infusión de Polivy administre un antihistamínico y un antipirético. La dosis inicial de Polivy debe administrarse en forma de infusión intravenosa de 90 minutos de duración. Se debe vigilar la aparición de reacciones relacionadas con la infusión durante esta y hasta al menos 90 minutos después de haber terminado de administrar la dosis inicial. Si el paciente tolera bien la primera infusión, la siguiente dosis de Polivy se puede administrar en infusión de 30 minutos de duración; se deberá vigilar al paciente durante la misma y hasta al menos 30 minutos después de haber terminado su administración.
Dosis diferidas u omitidas: Si se omite una dosis programada de Polivy, deberá administrarse lo antes posible, y se reajustará el esquema de administración para mantener el intervalo de 21 días entre dosis.
Modificaciones de la dosis: Si el paciente presenta una reacción relacionada con la infusión, se debe ralentizar o interrumpir la infusión de Polivy. Si la reacción es potencialmente mortal, se debe suspender la administración de Polivy de manera inmediata y permanente.
En la tabla 1 se resumen las modificaciones de la dosis en los pacientes que presenten una neuropatía periférica (NP).
|
Tabla 1. Modificaciones de la dosis de Polivy en los pacientes con neuropatía periférica |
|
|
Intensidad el día 1 de cualquier ciclo |
Modificación de la dosis |
|
Grado 2-3 |
Interrumpa la administración de Polivy hasta que la NP mejore a grado ≤1. Si la NP mejora a grado ≤1 el día 14 o antes, reanude la administración de Polivy reduciendo permanentemente la dosis a 1,4 mg/kg. Si ya se había reducido anteriormente la dosis a 1,4 mg/kg, suspenda definitivamente el tratamiento con Polivy. Si la NP no mejora a grado ≤1 el día 14 o antes, suspenda definitivamente el tratamiento con Polivy. |
|
Grado 4 |
Suspenda definitivamente el tratamiento con Polivy. |
En la tabla 2 se resumen las modificaciones de la dosis en los pacientes que presenten mielodepresión.
|
Tabla 2. Modificaciones de la dosis de Polivy, bendamustina y rituximab en los pacientes con mielodepresión |
|
|
Intensidad el día 1 de cualquier ciclo |
Modificación de la dosisa |
|
Neutropenia de grado 3-4 |
Interrumpa todo el tratamiento hasta que la cifra absoluta de neutrófilos (CAN) aumente a >1000 μl. Si la CAN aumenta a >1000 μl el día 7 o antes, reanude todo el tratamiento sin más reducciones de la dosis. Si la CAN aumenta a >1000 μl después del día 7: reanude todo el tratamiento reduciendo la dosis de bendamustina de 90 mg/m2 a 70 mg/m2 o de 70 mg/m2 a 50 mg/m2; si ya se había reducido la dosis de bendamustina a 50 mg/m2, suspenda definitivamente todo el tratamiento. |
|
Trombocitopenia de grado 3-4 |
Interrumpa todo el tratamiento hasta que la cifra de plaquetas aumente a >75 000 μl. Si la cifra de plaquetas aumenta a >75 000 μl el día 7 o antes, reanude todo el tratamiento sin más reducciones de la dosis. Si la cifra de plaquetas aumenta a >75 000 μl después del día 7: reanude todo el tratamiento reduciendo la dosis de bendamustina de 90 mg/m2 a 70 mg/m2 o de 70 mg/m2 a 50 mg/m2; si ya se había reducido la dosis de bendamustina a 50 mg/m2, suspenda definitivamente todo el tratamiento. |
|
a Si la causa primaria es el linfoma, puede que no haya que reducir la dosis de bendamustina. |
|
Pautas posológicas especiales: Uso en pediatría: No se han determinado la seguridad y la eficacia de Polivy en niños y adolescentes (menores de 18 años).
Uso en geriatría: No es preciso ajustar la dosis de Polivy en los pacientes de 65 años en adelante (v. Uso en geriatría y Farmacocinética en poblaciones especiales).
Disfunción renal: No es preciso ajustar la dosis de Polivy en los pacientes con un aclaramiento de la creatinina (ClCr) ≥30 ml/min. No se ha determinado una dosis recomendada en los pacientes con ClCr <30 ml/min (v. Disfunción renal y Farmacocinética en poblaciones especiales).
Disfunción hepática: No es preciso ajustar la dosis de Polivy en los pacientes con concentraciones de aspartato-transaminasa (AST) o alanina-transaminasa (ALT) de hasta 2,5 veces el límite superior de la normalidad (LSN) o concentraciones de bilirrubina total de hasta 1,5 veces el LSN. No se ha determinado una dosis recomendada en los pacientes con concentraciones de AST >2,5×LSN o ALT >2,5×LSN, o de bilirrubina total >1,5×LSN, o en los pacientes receptores de un trasplante hepático (v. Disfunción hepática y Farmacocinética en poblaciones especiales).
SOBREDOSIS: No hay experiencia de casos de sobredosis en ensayos clínicos en seres humanos. La dosis más alta estudiada hasta la fecha ha sido de 2,4 mg/kg administrados en infusión intravenosa. En caso de producirse una sobredosis, se debe interrumpir de inmediato la infusión y vigilar estrechamente al paciente.
PRESENTACIONES: Caja por 1 vial que contiene 140 mg de polatuzumab vedotina (Reg. San. No. INVIMA 2022MBT-0000046). Condición de venta: Venta bajo fórmula médica.
Mayor información:
PRODUCTOS ROCHE, S. A.
Bogotá, Colombia
CDS 1.0 - Dic 2018


