

SIMPONI
GOLIMUMAB
Inyectable
Jeringa(s) prellenada(s), Solución inyectable, 0.5 Mililitros
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA: SIMPONI® es un anticuerpo monoclonal IgG1k humano que tiene múltiples glicoformas, con una masa molecular esperada de entre 149802 y 151064 Dalton. SIMPONI® se produce en una línea celular recombinante cultivada con perfusión continua y se purifica mediante una serie de pasos que incluyen medidas para inactivar y eliminar virus.
SmartJect® pluma autoinyectable/precargada: Cada pluma autoinyectable/precargada de uso único de 0,5 ml contiene 50 mg de golimumab.
Cada VIAL de uso único contiene: 50 mg de golimumab por 4 mL (o 12.5 mg de golimumab por mL)
INDICACIONES:
Artritis reumatoide (AR): SIMPONI®, mediante administración subcutánea (SC), en combinación con metotrexato (MTX), está indicado para el tratamiento de artritis reumatoide activa, en pacientes adultos cuando la respuesta a los fármacos antirreumáticos modificadores de la enfermedad (FAMES), incluido el MTX, ha sido inadecuada. Inhibición de la progresión del daño estructural.
SIMPONI® IV, mediante administración intravenosa (IV), en combinación con metotrexato (MTX), está indicado para el tratamiento de artritis reumatoide activa, en pacientes adultos cuando la respuesta a los fármacos antirreumáticos modificadores de la enfermedad (FAMES), incluido el MTX, ha sido inadecuada. Inhibición de la progresión del daño estructural.
Artritis psoriásica (APs): SIMPONI®, mediante administración subcutánea, en combinación con MTX, está indicado para el tratamiento de artritis psoriásica activa, en pacientes adultos cuando la respuesta al tratamiento previo con fármacos antirreumáticos modificadores de la enfermedad (FAMES), no ha sido adecuada. Inhibición de la progresión del daño estructural.
SIMPONI® IV, mediante administración intravenosa, solo o en combinación con MTX, está indicado para:
• Reducir los signos y síntomas.
• Mejorar la función física.
• Inhibir la progresión del daño estructural.
• Mejorar la entesitis y la dactilitis.
• Mejorar la psoriasis y la enfermedad de psoriasis en uñas.
• Mejorar la calidad de vida relacionada con la salud de pacientes adultos con artritis psoriásica activa.
Espondilitis anquilosante (EA): SIMPONI®, mediante administración subcutánea, está indicado para el tratamiento de la espondilitis anquilosante activa, en pacientes adultos que han respondido de forma inadecuada al tratamiento convencional.
SIMPONI® IV, mediante administración administración intravenosa, está indicado para:
• Reducir los signos y síntomas.
• Mejorar la función física.
• Mejorar el rango de movimiento.
• Mejorar la calidad de vida relacionada con la salud de pacientes adultos con espondilitis anquilosante activa.
Espondiloartritis axial no radiográfica (nr Axial SpA): SIMPONI®, mediante administración subcutánea, está indicado en pacientes adultos con espondiloartritis axial activa no radiográfica severa con signos objetivos de inflamación, como se indica por la evidencia de proteína C-reactiva (PCR) elevada y/o resonancia magnética (MRI), que han tenido una respuesta inadecuada a, o son intolerantes a, medicamentos antiinflamatorios no esteroides (AINEs).
Colitis ulcerativa (CU): SIMPONI®, mediante administración subcutánea, está indicado para el tratamiento de colitis ulcerativa activa, de moderada a severa, en pacientes adultos que han respondido en forma inadecuada al tratamiento convencional.
DATOS FARMACÉUTICOS:
Lista de Excipientes:
Pluma autoinyectable/precargada:
L-histidina
L-histidina hidrocloruro
Polisorbato 80
Sorbitol
Agua para Inyección
Vial:
L – histidina
L-histidina hidrocloruro
Polisorbato 80
Sorbitol
Agua para Inyección
Incompatibilidades: No se han realizado estudios específicos de compatibilidad con otros fármacos.
Periodo de validez: Observe la fecha de caducidad en el envase exterior.
FORMA FARMACÉUTICA:
Pluma autoinyectable/precargada: Solución para inyección.
Vial: Solución concentrada para infusión.
PROPIEDADES FARMACOCINÉTICAS:
Absorción: Después de una administración única de SIMPONI® por vía SC a sujetos sanos o a pacientes con AR, el tiempo para alcanzar la concentración sérica máxima (™ax) osciló de 2 a 6 días. Una inyección SC de 50 mg de SIMPONI® en sujetos sanos causó una concentración sérica máxima (Cmax) media ± desviación estándar de 3.1 ± 1.4 mg/ml. La Cmax y el área bajo la curva (AUC) concentración-tiempo se incrementaron proporcionalmente con dosis por encima del rango de 50 a 400 mg después de una única administración por vía SC.
Después de una única inyección por vía SC de 100 mg en sujetos sanos, la absorción de SIMPONI® fue similar en la parte superior del brazo, el abdomen y el muslo, con una biodisponibilidad absoluta media del 51%. Debido a que la farmacocinética de SIMPONI® tras la administración SC de una dosis era casi proporcional a la dosis, es previsible que la biodisponibilidad absoluta de una dosis de 50 mg o 200 mg sea similar.
Después de una única administración por vía IV de 2 mg/kg de SIMPONI®, se observó una media de la Cmáx de 44.4 ±11.3 mg/mL en pacientes con AR.
Distribución: Después de una única administración IV, la media del volumen de distribución se estimó en 115 ± 19 mL/kg en sujetos sanos y 151 ± 61 mL/kg en pacientes con AR. El volumen de distribución de SIMPONI® indica que SIMPONI® se distribuye principalmente en el sistema circulatorio con limitada distribución extravascular.
Metabolismo: Se desconoce la vía metabólica exacta de SIMPONI®.
Eliminación: Después de una única administración IV, la depuración sistémica de SIMPONI® se estimó en 6.9 ± 2.0 mL/día/kg en sujetos sanos y 7.6 ± 2.0 mL/día/kg en pacientes con AR.
La vida media terminal fue consistente entre la administración IV y SC de SIMPONI®. La vida media terminal se estimó en 12 ± 3 días en sujetos sanos y se observó una vida media similar en pacientes con AR, APs, EA o CU.
Después de un tratamiento de 6 meses con SIMPONI® por administración subcutánea en pacientes con AR, el uso concomitante de metotrexato redujo la depuración aparente de SIMPONI® en 36%; sin embargo, después de la administración IV no se observó un efecto apreciable del metotrexato en la depuración de SIMPONI®. El análisis farmacocinético poblacional indicó que el uso concomitante de AINEs, corticosteroides orales o sulfasalacina no influenciaron la depuración aparente de SIMPONI® después de la administración SC.
En AR, APs y EA, el análisis farmacocinético poblacional indicó que el uso concomitante de MTX, AINEs, corticosteroides orales o sulfasalazina (SSZ) no influenció significativamente la depuración de golimumab después de la administración por vía IV.
El análisis farmacocinético poblacional mostró que después de la administración SC de SIMPONI®, los pacientes con niveles elevados de proteína C reactiva tendían a tener mayor depuración aparente de SIMPONI®. Los pacientes con niveles elevados de proteína C reactiva tuvieron más probabilidades de tener menores concentraciones séricas nadir de SIMPONI® después de la administración SC de SIMPONI®. En contraposición, el nivel de proteína C reactiva no mostró efecto en la depuración de SIMPONI® IV después de la administración IV de 2 mg/kg de SIMPONI® IV en la semanas 0, semana 4 y posteriormente cada 8 semanas.
Los pacientes que desarrollaron anticuerpos anti-SIMPONI® después de la administración SC o IV generalmente tuvieron bajas concentraciones séricas nadir de SIMPONI® en el estado estacionario.
Linealidad de la dosis: Después de una unica dosis por vía IV en pacientes con AR, SIMPONI® IV exhibió una farmacocinética proporcional a la dosis de aproximadamente un intervalo de dosis de 0.1 a 10.0 mg/kg. Después de una única dosis por vía SC en sujetos sanos, también se observó una farmacocinética proporcional a la dosis en un intervalo de dosis de 50 mg a 400 mg.
Dosis única frente a dosis múltiples: Los perfiles de concentración sérica-tiempo de SIMPONI® fueron generalmente predecibles después de la administración única o múltiple por vía SC o IV.
Cuando se administró 50 mg de SIMPONI® por vía SC a pacientes con AR, APs o EA cada 4 semanas, las concentraciones séricas alcanzaron el estado estacionario en la semana 12. Con el uso concomitante de metotrexato, el tratamiento con 50 mg de SIMPONI® por vía SC cada 4 semanas causó una media de la concentración sérica nadir en el estado estacionario de aproximadamente 0.4 ± 0.3 mg/mL en pacientes no tratados previamente con metotrexato con AR activa, aproximadamente 0.6 ± 0.4 mg/mL en pacientes con AR activa a pesar de la terapia con metotrexato, aproximadamente 0.5 ± 0.5 mg/mL en pacientes con AR activa previamente tratados con biológicos anti-TNFa, aproximadamente 0.5 ± 0.4 mg/mL en pacientes con APs activa y aproximadamente 0.8 ± 0.4 mg/mL en pacientes con EA. Los pacientes con AR, APs o EA que no recibieron concomitantemente metotrexato tuvieron concentraciones nadir de SIMPONI® en el estado estacionario aproximadamente 30% más bajas que aquellos que recibieron SIMPONI® con metotrexato. La concentración sérica media mínima en el estado estacionario en pacientes con APs fue 0.7 ± 0.6 mcg/mL. La concentración sérica media mínima en el estado estacionario en pacientes con AE fue 0.8 ± 0.6 mcg/mL.
La media de las concentraciones séricas nadir de golimumab en el estado estacionario en pacientes con nr Axial SpA fue similar a la observada en aquellos pacientes con EA después de la administración subcutánea de 50 mg de golimumab cada 4 semanas.
Cuando se administró 2 mg/kg de SIMPONI® IV por vía IV a pacientes con AR en la semana 0, semana 4 y posteriormente cada 8 semanas, las concentraciones séricas alcanzaron el estado estacionario en la semana 12. En pacientes con uso concomitante de metotrexato, el tratamiento con 2 mg/kg de SIMPONI® IV por vía IV cada 8 semanas causó una media de la concentración sérica nadir en el estado estacionario de aproximadamente 0.4 ± 0.4 mg/mL en pacientes con AR activa a pesar de la terapia con metotrexato.
Despues de la dosis de inducción de 200 mg y 100 mg de SIMPONI® por vía SC en la semana 0 y 2 respectivamente y de la dosis de mantenimiento de 100 mg de SIMPONI® por vía SC posteriormente cada 4 semanas en pacientes con CU, las concentraciones séricas de golimumab alcanzaron el estado estacionario aproximadamente 14 semanas después del inicio de la terapia. El tratamiento con 100 mg de SIMPONI® por vía SC cada 4 semanas durante el mantenimiento causó una media de la concentración sérica nadir en el estado estacionario de aproximadamente 1.8 ± 1.1 mg/mL. El uso concomitante de inmunomoduladores no tuvo ningún efecto aparante en los niveles nadir de golimumab en el estado estacinario cuando se administró 100 mg de SIMPONI® por vía SC cada 4 semanas en pacientes con CU.
Efecto del peso sobre la farmacocinética: El análisis farmacocinético poblacional mostró que hubo una tendencia hacia la depuración aparente mayor de SIMPONI® con el incremento del peso. Como un resultado, los pacientes con mayor peso tendían a tener menores concentraciones nadir de SIMPONI® en el estado estacionario después de la administración SC de una dosis de 50 mg o 100 mg; sin embargo, a través de las poblaciones de AR, APs y EA, un beneficio del tratamiento con SIMPONI® 50 mg se observó en todos los subgrupos por cuartiles de peso sin diferencias significativas en la eficacia clínica entre estos subgrupos. El tratamiento con el régimen de dosificación recomendado de SIMPONI® en pacientes con CU no causó diferencias significativas en la eficacia clínica entre los diferentes subgrupos de peso. Por lo tanto, no hay necesidad de ajustar la dosificación de SIMPONI® basados en el peso del paciente.
Después de la administración IV, los pacientes con mayor peso corporal tendían a tener concentraciones séricas ligeramente más elevadas de golimumab que los pacientes con menor peso cuando se administró golimumab basados en el peso corporal (mg/kg). Sin embargo, basados en el análisis farmacocinético poblacional, no hubo diferencias clínicamente relevantes en la exposición a golimumab después de la administración IV de 2 mg/kg de golimumab en pacientes en un rango de diferentes pesos corporales.
Poblaciones especiales:
Género: No se observaron diferencias farmacocinéticas relacionadas con el género con SIMPONI® después de la corrección del peso corporal de los pacientes.
Ancianos (≥ 65 años): Los parámetros farmacocinéticos de SIMPONI® no fueron influenciaddos por la edad en pacientes adultos. Los pacientes ≥ 65 años tuvieron una depuración aparente de SIMPONI® similar a la de los pacientes <65 años.
Origen étnico: No se observaron diferencias farmacocinéticas relacionadas con el origen étnico entre Caucásicos y Asiáticos.
Insuficiencia renal o hepática: No hay datos de farmacocinética disponibles en pacientes que tienen deterioro de la función renal o hepática.
Dosificación mensual: Los cinco estudios de Fase 3 por vía SC evaluaron la seguridad y la eficacia de SIMPONI® en un régimen de dosificación de cada 4 semanas; sin embargo, prospectivamente se permitió una ventana de 3 a 7 días. Los pacientes recibirían un total de 13 dosis en 1 año cuando SIMPONI® se administra cada 4 semanas en vez de 12 dosis cuando se administra una vez al mes. Esto resulta en una dosis mensual calculada de 54 mg frente a 50 mg, respectivamente y equivale a una diferencia de aproximadamente 8% en la exposición a golimumab.
PROPIEDADES FARMACODINÁMICAS:
Grupo farmacoterapéutico: Inhibidores del factor de necrosis tumoral alfa (TNF-a); código ATC: L04AB06.
Mecanismo de acción: Golimumab es un anticuerpo monoclonal humano que forma complejos estables de gran afinidad con ambas formas bioactivas del TNF humano, la soluble y la transmembrana, lo cual evita la unión del TNF a sus receptores. No se observó unión a otros ligandos de la superfamilia del TNF; en especial, golimumab no se une o neutraliza la linfotoxina humana. El TNFa se sintetiza principalmente por los monocitos activados, macrófagos y células T como una proteína transmembrana que se asocia a sí misma para formar el homotrimero bioactivo y es rápidamente liberado desde la superficie celular por proteólisis. La unión del TNF a los receptores p55 o p75 conlleva al agrupamiento de los dominios citoplasmáticos del receptor e inicia la señalización. Se ha identificado al TNF como una citoquina centinela esencial que es producida en respuesta a diversos estímulos y que posteriormente induce la respuesta inflamatoria a través de la activación de la apoptosis dependiente de caspasa y los factores de transcripción factor nuclear (NF)-kB y proteína activadora-1 (AP-1). El TNF también modula la respuesta inmunológica a través de su rol en la organización de células inmunológicas en los centros germinales. La expresión elevada del TNF se ha asociado a enfermedades inflamatorias crónicas como la artritis reumatoide, así como a las espondiloartropatías como artritis psoriásica, y espondilitis anquilosante y es un mediador importante de la inflamación articular y de las lesiones estructurales que son características de estas enfermedades.
CONTRAINDICACIONES: Hipersensibilidad al principio activo o a alguno de los excipientes. No se recomienda el uso en mujeres embarazadas y en lactancia. Niños menores de 18 años.
EMBARAZO, LACTANCIA Y FERTILIDAD:
Embarazo: Se realizó un estudio toxicológico del desarrollo embriofetal, en el cual se trataron monos cynomolgus preñadas durante el primer trimestre con dosis de golimumab de hasta 50 mg/kg dos veces por semana (más de 500 veces mayor a la dosis clínica propuesta de 50 mg cada 4 semanas, en términos de relación dosis/peso corporal). La concentración sérica materna media de los picos obtenida en este estudio (1576 µg/mL) fue más de 900 veces mayor que la mediana del valor de la Cmáx en el estado estacionario (1.71 µg/mL) después de una dosis subcutánea de 50 mg cada 4 semanas en pacientes con AR, APs y EA. Las muestras de sangre del cordón umbilical tomadas al final del segundo trimestre demostraron que los fetos estaban expuestos a golimumab durante la gestación. Las concentraciones séricas fetales fueron aproximadamente el 50% de las concentraciones séricas maternas. En este estudio, la exposición a golimumab in utero no causó defectos en el desarrollo del feto.
Se realizó un estudio sobre el desarrollo pre- y posnatal, en el cual se trataron con golimumab a monos cynomolgus preñadas durante el segundo y tercer trimestre y durante la lactancia. Se detectó golimumab en el suero de los neonatos desde el momento del parto y hasta seis meses después del parto. La concentración sérica materna media de los picos obtenida en este estudio (1482 mg/mL) fue más de 860 veces mayor que la mediana del valor de la Cmáx en el estado estacionario (1.71 µg/mL) después de una dosis subcutánea de 50 mg cada 4 semanas en pacientes con AR, APs y EA. La exposición a golimumab durante la gestación y durante el periodo postnatal no causó defectos en el desarrollo de las crías. Sin embargo, los estudios de desarrollo y reproducción en animales no siempre predicen la respuesta en humanos.
Golimumab atraviesa la placenta. Después del tratamiento con otro anticuerpo monoclonal bloqueador del TNF durante el embarazo, se ha detectado el anticuerpo hasta por 6 meses en el suero del recién nacido de una mujer tratada. Por consiguiente, estos infantes podrían tener un mayor riesgo de infección. No se recomienda la administración de vacunas de microorganismos vivos a infantes expuestos in utero a golimumab hasta 6 meses después de la última inyección de golimumab de la madre durante el embarazo (ver secciones Advertencias y precauciones e Interacciones).
Se desconoce si SIMPONI® puede causar daños al feto cuando se administra a una mujer embarazada o si puede afectar la capacidad reproductiva. Solo se debe administrar SIMPONI® a una mujer embarazada si es absolutamente necesario.
Lactancia: En el estudio del desarrollo pre- y posnatal en monos cynomolgus (ver sección Embarazo), en el cual se administró golimumab durante la gestación y la lactancia, se detectó golimumab en la leche materna en concentraciones de aproximadamente 350 veces menor que las concentraciones séricas maternas. Se desconoce si golimumab se excreta en la leche humana o si se absorbe sistémicamente después de la ingestión. Debido a que muchos medicamentos e inmunoglobulinas se excretan en la leche humana, y debido al potencial de SIMPONI® de producir reacciones adversas en los lactantes, se debe decidir si se descontinua la lactancia o se descontinua el tratamiento del medicamento, considerando la importancia del medicamento para la madre.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: No se han realizado estudios sobre los efectos en la capacidad para conducir o utilizar máquinas.
EFECTOS FARMACODINÁMICOS:
No clínicos: Se ha demostrado que la unión de golimumab al TNF humano neutraliza la expresión de la superficie celular, inducida por el TNF, de la molécula de adhesión selectina E, la molécula de adhesión de células vasculares (VCAM)-1 y la molécula de adhesión intercelular (ICAM)-1 en la superficie de las células endoteliales humanas. La secreción, inducida por el TNF, de las interleucinas (IL)-6, IL-8 y del factor estimulante de las colonias de granulocitos-macrófagos (GM-CSF) por las células endoteliales humanas también fue inhibida por golimumab. Del mismo modo que otros anticuerpos humanos IgG1, golimumab es capaz de unirse a los receptores Fc y de la activación del complemento. Sin embargo, no se observó lisis celular mediada por golimumab en monocitos humanos estimulados por el lipopolisacárido (LPS) con la adición de células del complemento o células efectoras. Adicionalmente, no se detectó apoptosis inducida por golimumab en células mononucleares de sangre periférica humana estimulada por el LPS. El efecto de golimumab in vivo se evaluó en un modelo de artritis experimental en ratón transgénico para el TNF humano. El tratamiento con golimumab causó un retraso estadísticamente significativo en la aparición de los síntomas clínicos en comparación con los ratones no tratados, así como una reducción significativa en la patología de las articulaciones.
Clínica:
Biomarcadores en AR, APs, EA y nr Axial SpA: En estudios clínicos de fase 3 por vía SC, fueron evaluados en el suero de pacientes con AR, APs y EA antes y después del inicio del tratamiento con SIMPONI® marcadores seleccionados de la inflamación y del metabolismo del hueso y cartílago. En el estudio GO-AHEAD, se evaluaron solo los niveles de la PCR. Sólo aquellos marcadores donde se observaron diferencias significativas (valores-p <0.05) en los pacientes tratados en el grupo combinado de SIMPONI® (50 mg y 100 mg, con o sin MTX) en comparación con los pacientes tratados con placebo/placebo + MTX se describen a continuación. No se ha establecido la relación entre los datos de los biomarcadores reportados y los mecanismos por los cuales golimumab ejerce sus efectos clínicos.
AR: En la primera evaluación (semana 4) después de la primera administración de SIMPONI®, SIMPONI® fue eficaz en modular los marcadores seleccionados de la inflamación y del metabolismo óseo en pacientes con AR activa. En pacientes con AR (AR activa a pesar del tratamiento con MTX, pacientes previamente tratados con otras terapias anti-TNFa, y pacientes sin tratamiento previo a MTX), el tratamiento con SIMPONI®/SIMPONI® + MTX causó una reducción significativa en los niveles de los marcadores inflamatorios seleccionados (tales como IL-6, TNFa, metalproteinasa de matriz (MMP)-3, ICAM-1 y factor de crecimiento endotelial vascular (VEGF)) ya desde la semana 4 en comparación con el tratamiento con placebo/placebo + MTX, y estos cambios se mantuvieron generalmente en las semanas 14/24. En los grupos de tratamiento con SIMPONI®/SIMPONI® + MTX, también se observó una mejoría de los niveles de la PCR en relación a los grupos de tratamiento con placebo/placebo + MTX. El tratamiento con SIMPONI®/SIMPONI® + MTX también causó una disminución significativa de los niveles del Factor Reumatoide (RF) en las semanas 14/24 en comparación con el tratamiento con placebo/placebo + MTX. En los pacientes con AR activa a pesar de la terapia con MTX, el tratamiento con SIMPONI® + MTX causó cambios significativos de los niveles de los marcadores seleccionados del metabolismo del hueso (incremento de los niveles de osteocalcina y propéptido N-terminal de procolágeno de tipo I (PINP) y disminución de los niveles de desoxi-piridinolina (DPD)) en la semana 4 en comparación con el placebo + MTX. Estos cambios de los biomarcadores son consistentes con una mejoría en el proceso de la enfermedad con inflamación reducida, crecimiento incrementado de los huesos y disminución de la resorción del hueso.
APs, EA y nr Axial SpA: En los estudios de fase 3 en APs y EA, en la primera evaluación (semana 4) después de la primera administración de SIMPONI®, hubo reducciones significativas de los marcadores seleccionados de la inflamación (IL-6, MMP-3, ICAM-1, VEGF, IL-8 [APs], TNFa, [EA]) y cambios significativos en los marcadores seleccionados del metabolismo óseo (incremento de la osteocalcina y PINP y disminución de los niveles de DPD) en comparación con el placebo. La disminución en IL-6, VEGF, IL-8 e I-CAM 1 se mantuvo en la semana 24 en los pacientes con APs tratados con SIMPONI®, y en los pacientes con EA, las reducciones se mantuvieron en la semana 24 para todos los marcadores inflamatorios, excepto TNFa. En los estudios en APs, EA y nr Axial SpA, en el grupo de tratamiento con SIMPONI® también se observó una mejoría de los niveles de la PCR en relación con el grupo de tratamiento con placebo. Estos cambios de los biomarcadores son consistentes con una mejoría en el proceso de la enfermedad con inflamación reducida, crecimiento incrementado de los huesos y disminución de la resorción del hueso.
Inmunogenicidad: Después de la administración SC en pacientes con AR, APs o EA, los anticuerpos contra SIMPONI®, casi todos neutralizados in vitro, fueron detectados en el 5% de los pacientes tratados con SIMPONI® con el método de inmunoensayo enzimático (EIA). Tasas similares fueron mostradas en todas las indicaciones reumatológicas. El tratamiento concomitante con MTX produjo una proporción menor de pacientes con anticuerpos a SIMPONI® que en los pacientes que recibieron SIMPONI® sin MTX (aproximadamente 3% frente al 8%, respectivamente).
Después de la administración SC en pacientes con nr Axial SpA, anticuerpos contra SIMPONI®, todos neutralizados in vitro, se detectaron en el 4% de los pacientes tratados con SIMPONI® hasta la semana 16 con el método de EIA.
Después de la administración SC en pacientes con CU, anticuerpos contra SIMPONI® se detectaron en el 2.7% de los pacientes tratados con SIMPONI® hasta la semana 54 con el método de EIA. Sesenta y ocho por ciento de los pacientes con resultado positivo a los anticuerpos habían neutralizando los anticuerpos in vitro. El tratamiento concomitante con inmunomoduladores (azatioprina, 6 mercaptopurina y MTX) produjo una menor proporción de pacientes con anticuerpos a SIMPONI® que los pacientes que recibieron SIMPONI® sin inmunomoduladores (1.3% frente al 3.4%, respectivamente).
Después de la administración IV de SIMPONI® IV en combinación con MTX en pacientes con AR, se detectaron anticuerpos contra golimumab en el 4.2% (39/922) de los pacientes tratados con golimumab hasta aproximadamente 1 año con el método de EIA. Todos los pacientes con resultado positivo a los anticuerpos contra golimumab habían neutralizado los anticuerpos in vitro.
El pequeño número de pacientes con resultado positivo a los anticuerpos contra SIMPONI® detectados con el método de EIA limita la capacidad de extraer conclusiones definitivas sobre la relación entre los anticuerpos a golimumab y las medidas de la eficacia y seguridad clínica.
Los datos reflejan el porcentaje de los pacientes cuyos resultados de la prueba se consideraron positivos para los anticuerpos contra SIMPONI® en el método de EIA.
Posteriormente se desarrolló un método de EIA tolerante al fármaco para la detección de anticuerpos contra golimumab. Este método es aproximadamente 16 veces más sensible que el método de EIA original con menos interferencia de golimumab en el suero. Debido a la mayor sensibilidad y a la tolerancia mejorada al fármaco, se esperaba que una mayor incidencia de anticuerpos contra golimumab sea detectada con el método de EIA tolerante al fármaco en comparación con el método de EIA original.
Después de la administración IV en pacientes con AR, APs o EA, se detectaron anticuerpos a SIMPONI® IV mediante el método EIA tolerante al fármaco en el 20% de los pacientes tratados con SIMPONI® IV (AR: 21%, APs: 19% y EA: 19%). Cuando se analizaron, aproximadamente un tercio de los anticuerpos a SIMPONI® IV fueron neutralizantes. El tratamiento concomitante con MTX produjo una proporción ligeramente menor de pacientes con anticuerpos a SIMPONI® IV que los pacientes que recibieron SIMPONI® IV sin MTX (aproximadamente 19% frente al 25%, respectivamente).
La mayor incidencia de los anticuerpos a golimumab con el método EIA tolerante al fármaco se debió principalmente a los anticuerpos de título bajo, los cuales no tuvieron un impacto aparente en las concentraciones, eficacia y seguridad del fármaco. Si bien los anticuerpos de título alto, los cuales fueron principalmente neutralizantes, pueden estar asociados con concentraciones más bajas del fármaco y eficacia disminuida, hubo pocos pacientes con títulos altos en los estudios por vía IV de APs y EA. El desarrollo de anticuerpos a SIMPONI® IV no evitó la respuesta clínica.
La incidencia observada de la respuesta positiva al anticuerpo puede estar influenciada por varios factores, incluyendo el manejo de la muestra, el momento de la recolección de la muestra, los medicamentos concomitantes, la enfermedad subyacente y particularmente la sensibilidad del ensayo. Por estas razones, la comparación de la incidencia de anticuerpos contra SIMPONI® con la incidencia de anticuerpos contra otros productos o resultados de diferentes ensayos puede ser confusa.
REACCIONES ADVERSAS: A través de esta sección, se presentan las reacciones adversas. Las reacciones adversas son eventos adversos que fueron considerados estar razonablemente relacionados con el uso de golimumab en base a la evaluación exhaustiva de la información disponible de los eventos adversos. No se puede establecer una relación causal con golimumab de forma segura en casos individuales. Además, debido a que los estudios clínicos se llevan a cabo en condiciones muy variables, las tasas de las reacciones adversas observadas en los estudios clínicos de un medicamento no pueden compararse directamente con las tasas de los estudios clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica clínica.
Están disponibles datos de seguridad, provenientes de estudios clínicos de fase 2 y 3, de 6161 pacientes tratados con golimumab, incluyendo 3090 pacientes con artritis reumatoide, 634 con artritis psoriásica, 768 con espondilitis anquilosante, 1245 con colitis ulcerativa, 231 con asma severo persistente y 193 con espondiloartritis axial no radiográfica activa (nr Axial SpA). En la tabla 1 se resumen las reacciones adversas observadas con golimumab.
En general, el perfil de seguridad general fue similar en pacientes que recibieron golimumab por las vías de administración subcutánea o intravenosa.
Dentro de la clasificación de órganos designada, las reacciones adversas se enumeran según su frecuencia, utilizando la siguiente convención:
Muy frecuente (≥ 1/10).
Frecuente (≥ 1/100, < 1/10).
Poco frecuente (≥ 1/1000, < 1/100).
Rara (≥ 1/10000, < 1/1000, incluyendo reportes aislados).
Desconocido (no puede ser estimado a partir de la información disponible).
|
Tabla 1: Resumen de las reacciones adversas en los estudios clínicos con golimumab |
|
|
Infecciones e infestaciones |
|
|
Muy frecuentes: |
Infección del tracto respiratorio superior (nasofaringitis, faringitis, laringitis y rinitis) |
|
Frecuentes: |
Infecciones bacterianas (como celulitis), infección del tracto respiratorio inferior (neumonía), infecciones víricas (como influenza y herpes), bronquitis, sinusitis, infecciones fúngicas superficiales, abscesos. |
|
Poco frecuentes: |
Sepsis, incluyendo shock séptico, pielonefritis |
|
Raras: |
Histoplasmosis, coccidioidomicosis, tuberculosis, infecciones oportunistas (infecciones fúngicas invasivas, bacterianas, por micobacterias atípicas y protozoos), reactivación de la hepatitis B, neumocistosis, artritis bacteriana, bursitis infecciosa |
|
Neoplasias benignas y malignas |
|
|
Raras: |
Linfoma, leucemia |
|
Desconocido: |
Neoplásias pediátricas |
|
Exploraciones complementarias |
|
|
Frecuentes: |
Alanina aminotransferasa incrementada, aspartato aminotransferasa incrementada |
|
Poco frecuentes: |
Recuento disminuido de neutrófilos |
|
Trastornos de la sangre y del sistema linfático |
|
|
Frecuentes: |
Leucopenia (incluyendo neutropenia), anemia. |
|
Poco frecuentes: |
Trombocitopenia, pancitopenia |
|
Trastornos del sistema inmunológico |
|
|
Muy frecuentes: |
Autoanticuerpos positivos, reacciones alérgicas no graves |
|
Trastornos del sistema nervioso |
|
|
Frecuentes: |
Mareos, parestesia |
|
Raras: |
Trastornos desmielinizantes (centrales y periféricos) |
|
Trastornos cardiacos |
|
|
Raras: |
Insuficiencia cardiaca congestiva (nueva aparición o empeoramiento) |
|
Trastornos vasculares |
|
|
Frecuentes: |
Hipertensión |
|
Raras: |
Vasculitis (sistémica) |
|
Trastornos respiratorios, torácicos y mediastínicos |
|
|
Poco frecuentes: |
Enfermedad pulmonar intersticial |
|
Trastornos gastrointestinales |
|
|
Poco frecuentes: |
Estreñimiento |
|
Trastornos de la piel y del tejido subcutáneo |
|
|
Frecuentes: |
Rash, alopecia |
|
Poco frecuentes: |
Psoriasis: nueva aparición o empeoramiento, palmar/plantar y pustular |
|
Raras: |
Vasculitis (cutánea) |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
|
|
Raras: |
Síndrome tipo Lupus |
|
Trastornos generales y alteraciones en el lugar de administración |
|
|
Frecuentes: |
Pirexia, reacción en el lugar de inyección (eritema en el lugar de inyección, urticaria, induración, dolor, hematoma, prurito, irritación, parestesia) |
Los datos descritos a continuación refleja las reacciones adversas en estudios clinicos de fase 2 y fase 3 por la vía subcutánea, excepto para reacciones de la administración y elevaciones de las enzimas hepáticas, las cuales incluyen datos de la vía subcutánea e intravenosa. A lo largo de esta sección, la mediana del tiempo de duración del seguimiento (aproximadamente 4 años) es generalmente presentada para todos los usos de golimumab. Donde el uso de golimumab se describe por dosis, la mediana del tiempo de duración del seguimiento varía (aproximadamente 2 años para la dosis de 50 mg y aproximadamente de 3 años para la dosis de 100 mg) ya que los pacientes pudieron cambiar entre las dosis.
Infecciones (ver sección Advertencias y precauciones): En el periodo controlado de los estudios pivotales, la infección del tracto respiratorio superior fue la reacción adversa más frecuentemente reportada en 12.6% de los pacientes en el grupo tratado con golimumab (incidencia por paciente-año: 0.61; IC del 95%: 0.55, 0.67) en comparación con el 11.0% de los pacientes control (incidencia por paciente-año: 0.55; IC del 95%: 0.46, 0.64). En el periodo controlado y no controlado de los estudios, con una mediana del tiempo de seguimiento de aproximadamente 4 años, la incidencia por paciente-año de las infecciones del tracto respiratorio superior fue de 0.35 casos (IC del 95%: 0.34, 0.36) para los pacientes tratados con golimumab.
En el periodo controlado de los estudios pivotales, se observaron infecciones en el 23.0% de los pacientes tratados con golimumab (incidencia por paciente-año: 1.32; IC del 95%: 1.23, 1.41) en comparación con el 20.2% de los pacientes control (incidencia por paciente-año: 1.22; IC del 95%: 1.09, 1.36). En el periodo controlado y no controlada de los estudios, con una mediana del tiempo de seguimiento de aproximadamente 4 años, la incidencia por paciente-año de las infecciones fue de 0.81 casos (IC del 95%: 0.79, 0.83) para los pacientes tratados con golimumab.
Se observaron infecciones graves en pacientes tratados con golimumab incluyendo sepsis, neumonía, celulitis, abscesos, infecciones oportunistas y tuberculosis. En el período controlado de los estudios en AR, APs, colitis ulcerativa, EA y nr Axial SpA se observaron infecciones graves en el 1.2% de los pacientes tratados con golimumab y en el 1.2% de los pacientes control. La incidencia de infecciones graves por paciente-año de seguimiento en el periodo controlado de los estudios en RA, APs, EA, nr Axial SpA fue 0.07; IC del 95%: 0.05, 0.11 para el grupo tratado con 100 mg de golimumab, 0.03; IC del 95%: 0.01, 0.06 para el grupo tratado con 50 mg de golimumab y 0.04; IC del 95%: 0.02, 0.07 para el grupo placebo. En el período controlado de los estudios en CU con inducción de golimumab, se observaron infecciones graves en el 0.8% de los pacientes tratados con golimumab en comparación con el 1.5% de los pacientes tratados con el control. En el periodo controlado y no controlado de los estudios pivotales con una mediana de seguimiento de hasta 3 años, hubo una mayor incidencia de infecciones graves, incluyendo infecciones oportunistas y tuberculosis, en los pacientes que recibieron 100 mg de golimumab en comparación con los pacientes que recibieron 50 mg de golimumab. La incidencia por paciente-año de todas las infecciones graves fue de 0.04; IC del 95%: 0.04, 0.05 en los pacientes que recibieron 100 mg de golimumab y 0.03; IC del 95%: 0.02, 0.03 en pacientes que recibieron 50 mg de golimumab. Estos resultados pueden malinterpretarse por el diseño de los estudios pivotales y la diferente duración del seguimiento entre los grupos tratados.
Neoplasias malignas (ver sección Advertencias y precauciones):
Linfoma: La incidencia de linfoma en los pacientes tratados con golimumab durante los estudios pivotales, fue mayor que la esperada en la población general. En el periodo controlado y no controlado de estos estudios, con una mediana de seguimiento de hasta 3 años, se observó una mayor incidencia de linfoma en los pacientes que recibieron 100 mg de golimumab en comparación con los pacientes que recibieron 50 mg de golimumab. Estos resultados pueden malinterpretarse por del número pequeño de casos, el diseño de los estudios en fase 3 y la diferente duración del seguimiento entre los grupos tratados. La mayoría de linfomas ocurrieron en el Estudio 2 en AR, que incluyó pacientes previamente expuestos a agentes anti-TNF siendo su enfermedad de mayor duración y más refractaria.
Neoplasias malignas distintas al linfoma: En los periodos controlados de los estudios pivotales, la incidencia de neoplasias malignas distintas al linfoma (excluyendo cáncer de piel no melanoma) fue similar entre los grupos tratados con golimumab y con el control. Durante aproximadamente 4 años de seguimiento, la incidencia de neoplasias malignas distintas al linfoma (excluyendo cáncer de piel no melanoma) fue similar al de la población en general.
En un ensayo clínico exploratorio donde se incluyó a pacientes con asma severa persistente, más pacientes tratados con golimumab presentaron neoplasias malignas en comparación con los pacientes control. Se desconoce la significancia de estos hallazgos en la población con asma.
Se desconoce el rol potencial del tratamiento con bloqueadores del TNF en el desarrollo de las neoplasias malignas.
Trastornos desmielinizantes (ver sección Advertencias y precauciones): En los periodos controlados y no controlados de los estudios pivotales, con una mediana de seguimiento de hasta 3 años, se observó una mayor incidencia de desmielinización en los pacientes que recibieron 100 mg de golimumab en comparación con los pacientes que recibieron 50 mg de golimumab. Estos resultados pueden malinterpretarse por el número pequeño de casos, el diseño de los estudios pivotales y la diferente duración del seguimiento entre los grupos tratados.
Incremento de las enzimas hepáticas: En los periodos controlados de los estudios pivotales en AR y APs se produjo un leve incremento de la ALT [> 1 y < 3 x límite superior de la normalidad (LSN)] en una proporción similar en los pacientes tratados con golimumab y en los pacientes control (22.1% al 27.4% de los pacientes); en los estudios en EA y nr Axial SpA, más pacientes tratados con golimumab (26.9%) que los pacientes control (10.6%) presentaron leve incremento de la ALT. En los periodos controlados y no controlados de los estudios pivotales en AR y APs, con una mediana de seguimiento de aproximadamente 5 años, la incidencia del leve incremento de la ALT fue similar en los pacientes tratados con golimumab y en los pacientes control.
En el periodo controlado de los estudios pivotales en CU con inducción de golimumab, se produjo leve incremento de la ALT (> 1 y < 3 x LSN) en similar proporción en los pacientes tratados con golimumab y en los pacientes control (7.8% a 6.9%, respectivamente). En los periodos controlados y no controlados de los estudios pivotales en CU con un mediana de seguimiento de aproximadamente 2 años, la proporción de pacientes con leve incremento de la ALT fue 24.7% en los pacientes que recibieron golimumab.
En el periodo controlado de los estudios pivotales en AR y EA, los incrementos de la ALT ≥ 5 x LSN fueron poco frecuentes y se observaron más en pacientes tratados con golimumab (0.4% al 0.9%) que en los pacientes control (0.0%). Esta tendencia no fue observada en la población con APs. En los periodos controlados y no controlados de los estudios pivotales en AR, APs y EA, con una mediana de seguimiento de hasta 5 años, la incidencia del incremento de la ALT ≥ 5 x LSN fue similar en los pacientes tratados con golimumab y en los pacientes control. La mayoría de estos incrementos fueron asintomáticos. No se reportaron casos en los períodos controlados y no controlados del estudio en nr Axial SpA (hasta por 1 año).
En los periodos controlados de los estudios pivotales en CU con inducción de golimumab, se produjeron incrementos de la ALT ≥ 5 x LSN en similar proporción en los pacientes tratados con golimumab en comparación con los pacientes tratados con placebo (0.3% a 1.0%, respectivamente). En los periodos controlados y no controlados de los estudios pivotales en CU con una mediana de seguimiento de aproximadamente 2 años, la proporción de pacientes con incrementos de la ALT ≥ 5 x LSN fue 0.8% en los pacientes que recibieron golimumab.
En los estudios pivotales por vía IV, los incrementos de las enzimas hepáticas fueron comparables a los observados en los estudios por vía subcutánea con las siguientes excepciones:
En el período controlado del estudio pivotal por vía IV de APs se observaron elevaciones leves de ALT (> 1 y < 3 x LSN) en mayor número en los pacientes tratados con golimumab (34%) que en los pacientes control (26%).
En el período controlado del estudio pivotal por vía IV de APs se observaron elevaciones de ALT ≥ 3 y < 5 LSN en mayor número en los pacientes tratados con golimumab (2.9%) que en los pacientes control (0.4%).
En el período controlado del estudio pivotal por vía intravenosa de APs se observaron elevaciones de ALT ≥ 5 x LSN en mayor número en los pacientes tratados con golimumab (1.7%) que en los pacientes control (0.4%).
Reacciones en el lugar de la inyección: En los periodos controlados de los estudios pivotales, el 5.4% de los pacientes tratados con golimumab tuvieron reacciones en el lugar de la inyección en comparación con el 2.0% de los pacientes tratados con el control. La mayoría de las reacciones en el lugar de la inyección fueron leves y moderadas y la manifestación más frecuente fue eritema en el lugar de la inyección.
En los periodos controlados de los estudios pivotales por vía IV el 0.2% de los sujetos tratados con placebo y el 2.8% de los sujetos tratados con golimumab tuvieron una reacción debido a la infusión. Las reacciones más comunes a la infusión fueron erupción y dolor de cabeza. No se reportaron reacciones graves por la infusión.
En los estudios controlados de fase 2 y/o 3 en AR, APs, EA, nr Axial SpA, asma severo persistente y en estudios de fase 2/3 en CU, ningún paciente tratado con golimumab desarrolló reacciones anafilácticas que se consideren relacionadas a golimumab.
Anticuerpos antinucleares (ANA)/anticuerpos anti-ADN de doble cadena (dsDNA): En los periodos controlados y no controlados de los estudios pivotales hasta el año de seguimiento, el 3.5% de los pacientes tratados con golimumab y el 2.3% de los pacientes control tuvieron resultados positivos para ANA de nueva aparición (títulos de 1:160 o mayor). La frecuencia de los anticuerpos anti-dsDNA al año de seguimiento en los pacientes que en el estado basal dieron resultados negativos para anti-dsDNA fue 1.1% (ver sección Advertencias y precauciones).
Experiencia posterior a la comercialización: Las frecuencias que se indican a continuación reflejan la tasa de reporte de reacciones adversas a partir de la experiencia mundial posterior a la comercialización con golimumab. No se pueden realizar estimaciones precisas de la incidencia debido al reporte voluntario de una población de tamaño incierto. Estas reacciones adversas se clasifican según frecuencia, utilizando la siguiente convención: Muy frecuente (≥ 1/10), frecuente (≥ 1/100 y < 1/10), poco frecuente (≥ 1/1000 y < 1/100), rara (≥ 1/10000 y < 1/1000), muy rara (< 1/10000, incluyendo reportes aislados), desconocido (no se puede estimar a partir de la información disponible).
|
Tabla 2: Reacciones adversas a golimumab posteriores a la comercialización |
||
|
Sistema de clasificación de órganos |
Reacción adversa |
Frecuencia |
|
Trastornos generales y condiciones en el lugar de la administración |
Reacción relacionada a la infusión |
Común |
|
Neoplasia benigno y maligno |
Melanoma |
Rara |
|
Carcinoma de células de Merkel Linfoma de células T hepatoesplénico* |
Desconocido |
|
|
Trastornos del sistema inmunológico |
Reacciones de hipersensibilidad sistémica grave (incluyendo reacción anafiláctica) |
Rara |
|
Sarcoidosis |
||
|
Trastornos de la piel y del tejido subcutáneo |
Reacciones cutáneas ampollosas |
Poco frecuente |
|
Exfoliación de la piel |
Rara |
|
|
*Observado con otros agentes bloqueadores del TNF |
||
INTERACCIONES: No se han realizado estudios específicos de interacción farmacológica con SIMPONI®.
Uso concomitante de SIMPONI® con otras terapias biológicas: No se recomienda combinar SIMPONI® con otras terapias biológicas utilizadas para tratar las mismas afecciones que SIMPONI®, incluyendo anakinra y abatacept (ver sección Advertencias y precauciones).
Vacunas de microorganismos vivos/Agentes infecciosos terapéuticos: No se deben administrar vacunas de microorganismos vivos concomitantemente con SIMPONI® (ver sección Advertencias y precauciones).
No se debe administrar los agentes infecciosos terapéuticos concomitantemente con SIMPONI® (ver sección Advertencias y precauciones).
Metotrexato: No se ha observado efectos significativos de metotrexato sobre la depuración de SIMPONI® IV administrado por vía intravenosa. Después de la administración subcutánea, el uso concomitante de metotrexato causó un incremento de las concentraciones mínimas en estado estacionario de SIMPONI® en pacientes con AR, APs o EA. Sin embargo, los datos no sugieren la necesidad de ajustar la dosis de SIMPONI® ni de metotrexato (ver sección Propiedades farmacocinéticas).
ESTUDIOS CLÍNICOS:
Artritis reumatoide:
Diseños de estudio: La eficacia y seguridad de SIMPONI® fueron evaluados en cuatro estudios multicéntricos, aleatorizados, doble ciego, controlado con placebo (3 por vía subcutánea [Estudio 1 en AR , Estudio 2 en AR y Estudio 3 en AR] y 1 por vía intravenosa [Estudio 1 en AR por vía IV]) en más de 2100 pacientes ≥ 18 años de edad con AR activa de moderada a severa diagnosticada según los criterios del American College of Rheumatology (ACR) por lo menos tres meses antes de ser diagnosticados. En el Estudio 1, 2 y 3 en AR los pacientes tenían por lo menos 4 articualciones dolorosas y 4 articulaciones inflamadas. SIMPONI® se administró por vía subcutánea en dosis de 50 mg o 100 mg, con o sin MTX, cada 4 semanas. En el Estudio 1 en AR por vía IV, los pacientes tenían por lo menos 6 articulaciones dolorosas y 6 articulaciones inflamadas. SIMPONI® IV se administró en una infusión intravenosa a dosis de 2 mg/kg con MTX en la semana 0 y 4 y posteriormente cada 8 semanas. Los criterios clínicos de evaluación incluyeron evaluaciones de la respuesta ACR, la Escala de Actividad de la Enfermedad (DAS 28), el Cuestionario de Evaluación de la Salud (HAQ) y calidad de vida relacionada con la salud.
Estudio 1 de AR: Artritis reumatoide activa a pesar del tratamiento con MTX: En el estudio 1 en AR (GO-FORWARD) se evaluaron 444 pacientes con AR activa a pesar de recibir una dosis estable de por lo menos 15 mg/semana de MTX y que no habían sido tratados previamente con un agente anti-TNF. Los pacientes fueron aleatorizados para recibir placebo + MTX (N=133), SIMPONI® 50 mg + MTX (N=89), SIMPONI® 100 mg + MTX (N=89) o monoterapia de SIMPONI® 100 mg + placebo (N=133). Los criterios de evaluación co-principales fueron el porcentaje de pacientes que alcanzaron respuesta ACR 20 en la semana 14 y la mejoría desde el estado basal del HAQ en la semana 24. Los principales criterios de evaluación secundaria incluyeron cambios desde el estado basal de la puntuación van der Heijde Sharp (vdH-S) en la semana 24, respuesta DAS28 (usando PCR) en la semana 14, respuesta ACR 20 en la semana 24 y mejoría desde el estado basal del HAQ en la semana 14. Todos los pacientes que recibieron placebo + MTX recibieron SIMPONI® 50 mg + MTX después de la semana 24, pero el ensayo permaneció doble ciego hasta cuando todos los pacientes completaron 52 semanas de tratamiento. A la semana 52, los pacientes ingresaron a la fase de extensión a largo plazo en la cual los pacientes continuaron el tratamiento con SIMPONI® 50 mg + MTX, SIMPONI® 100 mg + MTX o SIMPONI® 100 mg en monoterapia. Después de que el último paciente completó la visita en la semana 52 y el ensayo fue dado a conocer, a los pacientes que recibieron SIMPONI® 50 mg se les podía incrementar la dosis a 100 mg a discreción del investigador, y a los pacientes que recibieron SIMPONI® 100 mg en monoterapia se les podía añadir MTX. Los datos de la eficacia se recogieron y analizaron hasta la semana 256.
Estudio 2 de AR: Artritis reumatoide activa, previamente tratada con agente(s) anti-TNFa: En el estudio 2 en AR (GO-AFTER) se evaluaron 445 pacientes que fueron previamente tratados con uno o más de los siguientes agentes anti-TNF: adalimumab, etanercept o infliximab. Las razones para descontinuar los tratamientos anti-TNF previos incluyeron: falta de eficacia (59%), intolerancia (17%) y/u otras razones diferentes a la seguridad o eficacia (39%). Los pacientes fueron aleatorizados para recibir placebo (N=150), SIMPONI® 50 mg (N=147) y SIMPONI® 100 mg (N=148). Se les permitió a los pacientes continuar con un tratamiento concomitante de FAMES con MTX, sulfasalazina y/o hidroxicloroquina durante el estudio. El principal criterio de evaluación fue el porcentaje de pacientes que alcanzaron respuesta ACR 20 en la semana 14. Los principales criterios de evaluación secundaria incluyeron respuesta ACR 50 en la semana 14, respuesta DAS28 (usando PCR) en la semana 14, respuesta ACR 20 en la semana 24 y mejoría desde el estado basal de la puntuación del HAQ en la semana 24. En la semana 24, los pacientes ingresaron a la fase de extensión a largo plazo en la cual los pacientes continuaron el tratamiento con SIMPONI® 50 mg o SIMPONI® 100 mg; todos los pacientes que recibieron placebo comenzaron a recibir SIMPONI® 50 mg en la semana 24. Después que el último paciente completó la visita en la semana 24 y el ensayo fue dado a conocer, a los pacientes que recibieron SIMPONI® 50 mg se les podía incrementar la dosis a 100 mg a discreción del investigador. Los datos de la eficacia se recogieron y analizaron hasta la semana 256.
Estudio 3 de AR : Artritis reumatoide activa, sin tratamiento previo con MTX: En el Estudio 3 en AR (GO-BEFORE) se evaluaron 637 pacientes con AR activa sin tratamiento previo con MTX y que no habían sido tratados con un agente anti-TNF. Los pacientes fueron aleatorizados para recibir placebo + MTX (N=160), SIMPONI® 50 mg + MTX (N=159), SIMPONI® 100 mg + MTX (N=159) o monoterapia de SIMPONI® 100 mg + placebo (N=159). En los pacientes que recibieron MTX activo, el MTX se administró a una dosis de 10 mg/semana comenzando en la semana 0 e incrementando la dosis a 20 mg/semana en la semana 8. Los criterios de evaluación co-principales fueron el porcentaje de pacientes que alcanzaron respuesta ACR 50 en la semana 24 y el cambios desde el estado basal de la puntuación vdH-S en la semana 52. Los principales criterios de evaluación secundaria incluyeron el cambio desde el estado basal del HAQ en la semana 52, respuesta ACR 20 en la semana 24, el cambio desde el estado basal de la puntuación vdH-S en la semana 52 en pacientes con PCR anormal en el estado basal y el porcentaje de pacientes con CRP anormal en el estado basal que alcanzarón una respuesta ACR 50 en la semana 24. En la semana 52, los pacientes que recibieron placebo + MTX que tenian por lo menos 1 articulación dolorosa o articulación inflamada empezaron a recibir SIMPONI® 50 mg + MTX. Los pacientes que no tenían articulación dolorosa o articulación inflamada en la semana 52 continuaron recibiendo el placebo + MTX después de la semana 52. En la semana 52, los pacientes ingresaron a la fase de extensión a largo plazo en la cual la mayoría de los pacientes continuaron con SIMPONI® 50 mg + MTX, SIMPONI® 100 mg + MTX o SIMPONI® 100 mg en monoterapia. El ensayo permaneció doble ciego hasta que todos los pacientes completaron las 52 semanas de tratamiento. Después de que el último paciente completó la visita en la semana 52 y el ensayo fue dado a conocer, a los pacientes que recibieron SIMPONI® 50 mg se les podía incrementar la dosis a 100 mg a discreción del investigador, y a los pacientes que recibieron SIMPONI® 100 mg en monoterapia se les podía añadir MTX. Los datos de eficacia se recogieron y analizaron hasta la semana 256.
Estudio 1 de AR por vía IV: Artritis reumatoide activa a pesar del tratamiento con MTX: En el Estudio 1 en AR por vía IV (GO-FURTHER) se evaluaron 592 pacientes con AR activa a pesar de la terapia concomitante con MTX. Los pacientes fueron aleatorizados para recibir SIMPONI® IV 2 mg/kg por vía intravenosa (n=395) o placebo por vía intravenosa (solución salina) (N=197) en la semana 0, semana 4 y posteriormente cada 8 semanas además de su dosis semanal de mantenimiento de MTX. Todos los pacientes que recibieron el placebo por via IV + MTX recibieron SIMPONI® I.V. 2 mg/kg por vía IV + MTX después de la semana 24, pero el ensayo permaneció doble ciego hasta las 52 semanas de tratamiento. El principal criterio de evaluación fue el porcentaje de pacientes que alcanzaron respuesta ACR 20 en la semana 14. Los principales criterios de evaluación secundaria incluyeron respuesta DAS28 (usando PCR) y cambio desde el estado basal del HAQ-DI en la semana 14 así como respuesta ACR 50 y cambio desde el estado basal de la puntuación vdH-S en la semana 24. Otros criterios de evaluación previamente especificadas incluyeron mejoría de los componentes ACR, respuesta ACR en el tiempo, mejoría del funcionamiento físico y la calidad de vida relacionada con la salud así como medidas de economía sanitaria.
Reducción de los signos y síntomas: En general, no se apreciaron diferencias clínicamente significativas en las mediciones de la eficacia entre los regímenes de dosificación de SIMPONI® 50 mg y 100 mg en cada uno de los estudios de fase 3 en AR hasta la semana 104 en el Estudio 1 y el Estudio 3 en AR, y hasta la semana 24 en el Estudio 2 en AR. En los Estudios 1, 2 y 3 en AR, mediante el diseño del estudio, los pacientes en la extensión de largo plazo podían cambiar entre las dosis de SIMPONI® de 50 mg y 100 mg a discreción del médico del estudio.
Estudio 1 de AR: El tratamiento con SIMPONI® en pacientes con AR activa a pesar del tratamiento con MTX causó una mejoría de los signos y síntomas como se demostró con el porcentaje de pacientes que alcanzaron una respuesta ACR 20 en la semana 14. Un porcentaje significativamente mayor de pacientes alcanzaron una respuesta ACR 20 en el grupo combinado de SIMPONI® + MTX y en los grupos de dosificación individual de SIMPONI® + MTX que en el grupo del placebo + MTX (p ≤ 0.001 para todas las comparaciones). En el grupo de SIMPONI® 50 mg + MTX, el 55% de los pacientes alcanzaron una respuesta ACR 20 en comparación con el 33% en el grupo del placebo + MTX en la semana 14. El porcentaje de pacientes que alcanzaron una respuesta ACR 20 en la semana 24 también fue significativamente mayor en los pacientes que recibieron SIMPONI® 50 mg + MTX en comparación con el grupo del placebo + MTX (60% en comparación con el 28%, respectivamente) (Tabla 3).
Cuando se consideraron las respuestas ACR 20 a través del tiempo, se observó una mejoría en la primera evaluación (Semana 4) después de la primera administración de SIMPONI® 50 mg + MTX y se mantuvo hasta la semana 24 (Figura 1).
Figura 1: Estudio 1 de AR: Porcentaje de pacientes que alcanzarón respuesta ACR 20 hasta la semana 24; pacientes aleatorizados en los grupos con dosis de placebo + MTX y SIMPONI® de 50 mg + MTX
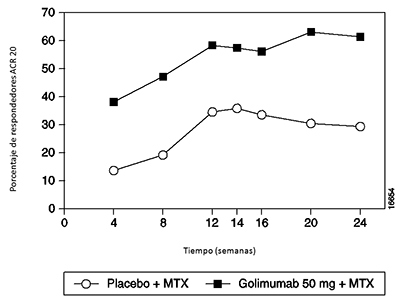
El porcentaje de pacientes que alcanzaron respuestas ACR 50 y ACR 70 también fue mayor en el grupo de SIMPONI® 50 mg + MTX que en el grupo del placebo + MTX (Tabla 3). El porcentaje de pacientes que alcanzaron una respuesta ACR 50 en los grupos de SIMPONI® 50 mg + MTX y del placebo + MTX, fue de 35% y 10%, respectivamente en la semana 14, y 37% y 14%, respectivamente en la semana 24 (p < 0.001 para todas las comparaciones). El porcentaje de pacientes que alcanzaron una respuesta ACR 70 en los grupos de SIMPONI® 50 mg + MTX y del placebo + MTX fue 14% y 4%, respectivamente en la semana 14 (p = 0.008) y 20% y 5%, respectivamente en la semana 24 (p < 0.001).
Las tasas de las respuestas ACR 20, ACR 50 o ACR 70 se mantuvieron hasta la semana 104. Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de respuestas ACR 20, ACR 50 o ACR 70 desde la semana 104 hasta la semana 256.
El porcentaje de pacientes que alcanzaron una respuesta DAS28 (usando PCR) fue significativamente mayor en aquellos pacientes tratados con SIMPONI® 50 mg + MTX en comparación con aquellos que recibieron el placebo + MTX en la semana 14 (72% en comparación con 50%; p=0.001) y en la semana 24 (73% en comparación con 42%; p<0.001).
El porcentaje de pacientes que alcanzaron remisión DAS28 (usando PCR) fue significativamente mayor en aquellos pacientes tratados con SIMPONI® 50 mg + MTX en comparación con los que recibieron el placebo + MTX en la semana 14 (27% en comparación con 5%; p<0.001) y en la semana 24 (27% en comparación con 7%; p<0.001). De igual manera, se observaron resultados estadísticamente significativos cuando se evaluó la respuesta y la remisión DAS28 [usando tasa de sedimentación de eritrocitos (ESR)].
|
Tabla 3: Estudio 1 de AR: Porcentaje de pacientes con AR con respuesta ACR, respuesta y remisión DAS en la semana 14 y la semana 24 |
||||||||||||||||||
|
Placebo + MTX (N= 133)a |
SIMPONI® 50 mg + MTX* (N = 89)a |
|||||||||||||||||
|
ACR 20 (% de respondedores) |
||||||||||||||||||
|
Semana 14 |
33% |
55% |
||||||||||||||||
|
Semana 24 |
28% |
60% |
||||||||||||||||
|
ACR 50 (% de respondedores) |
||||||||||||||||||
|
Semana 14 |
10% |
35% |
||||||||||||||||
|
Semana 24 |
14% |
37% |
||||||||||||||||
|
ACR 70 (% de respondedores) |
||||||||||||||||||
|
Semana 14 |
4% |
14% |
||||||||||||||||
|
Semana 24 |
5% |
20% |
||||||||||||||||
|
Respuesta DAS28 (usando PCR) (% de respondedores) |
||||||||||||||||||
|
Semana 14 |
50% |
72% |
||||||||||||||||
|
Semana 24 |
42% |
73% |
||||||||||||||||
|
Remisión DAS28 (usando PCR) (% en remitentes) |
||||||||||||||||||
|
Semana 14 |
5% |
27% |
||||||||||||||||
|
Semana 24 |
7% |
27% |
||||||||||||||||
|
Respuesta DAS28 (usando ESR) (% de respondedores) |
||||||||||||||||||
|
Semana 14 |
44% |
71% |
||||||||||||||||
|
Semana 24 |
42% |
72% |
||||||||||||||||
|
Remisión DAS28 (usando ESR) (% en remitentes) |
||||||||||||||||||
|
Semana 14 |
2% |
16% |
||||||||||||||||
|
Semana 24 |
6% |
20% |
||||||||||||||||
|
* p ≤ 0.001 para todas las comparaciones con excepción de la respuesta ACR 70 en la semana 14, donde p = 0.008. a N refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar en el punto cronológico. Una respuesta ACR 20 (Felson et al, 1995) se definió como: 1. ≥ 20% de mejoría en el recuento de articulaciones inflamadas (66 articulaciones) y recuento de articulaciones dolorosas (68 articulaciones); 2. ≥ 20% de mejoría en 3 de las siguientes cinco evaluaciones: • Evaluación de dolor del paciente en una escala VAS (escala analógica visual) de 0-10 cm (de ausencia de dolor al peor dolor posible). • Evaluación general del paciente de la actividad de la enfermedad en una escala VAS de 0-10 cm (de muy bien a muy mal). • Evaluación general del médico de la actividad de la enfermedad en una escala VAS de 0-10 cm (ausencia de artritis activa a artritis extremadamente activa). • Evaluación del funcionamiento físico del paciente medido por el HAQ en una escala de 0 a 3 (sin ninguna dificultad a incapacidad) • PCR. Una respuesta ACR 50 o ACR 70 se definió como ≥ 50% o ≥ 70% de mejoría en los anteriores numerales 1 y 2. La DAS28 (usando PCR) se derivó usando evaluaciones del recuento de 28 articulaciones inflamadas (inflamadas 28) y el recuento de 28 articulaciones dolorosas/sensibles (dolorosas 28), evaluación general del paciente de la actividad de la enfermedad (GH) usando la fórmula, combinada con PCR o ESR: DAS28=0.56 (raíz cuadrada(dolorosa 28)) + 0.28(raíz cuadrada(inflamada 28)) + 0.36(Ln (PCR+1)) [o 0.70)Ln(ESR)] + 0.014 (GH) + 0.96 (van der Linden, 2004; van Riel, 2000). La respuesta DAS28 en una visita se basó en la puntuación DAS28 vigente así como la mejoría desde el estado basal de la puntuación DAS28. Los pacientes se clasificaron como: Mejoría de la DAS28 desde el estado basal:
Los respondedores DAS28 incluyen pacientes con respuesta moderada o buena. Los remitentes DAS28 incluyen pacientes con un valor DAS28 de < 2.6 en una visita. |
||||||||||||||||||
La proporción de los pacientes tratados con SIMPONI® que alcanzaron respuesta o remisión DAS28 (usando PCR) en la semana 24 se mantuvo hasta la semana 52 y la semana 104. Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de la respuesta o remisión DAS28 (usando PCR) desde la semana 104 hasta la semana 256.
El tratamiento con SIMPONI® 50 mg + MTX también causó una mejoría significativamente mayor en cada componente ACR en comparación con el tratamiento con el placebo + MTX (Tabla 4). El recuento de articulaciones inflamadas en los grupos de SIMPONI® 50 mg + MTX y el placebo + MTX mejoró en 62% y 38%, respectivamente, en la semana 14, y 72% y 32%, respectivamente, en la semana 24. La mejoría en el recuento de las articulaciones dolorosas fue 60% en comparación con 30% en la semana 14 y 62% en comparación con 21% en la semana 24 en los grupos de SIMPONI® 50 mg + MTX y el placebo + MTX, respectivamente. Las evaluaciones de los pacientes y los médicos y la puntuación del HAQ también mejoraron significativamente para SIMPONI® 50 mg + MTX en comparación con el placebo + MTX en la semanas 14 y la semana 24. Para SIMPONI® 50 mg + MTX, hubo un 44% de mejoría de la PCR en comparación con 2% de mejoría con el placebo + MTX en la semana 14 y 39% en comparación con 0% en la semana 24.
|
Tabla 4: Estudio 1 de AR: Porcentaje de mejoría de los componentes ACR en la semana 14 y la semana 24 en pacientes aleatorizados |
||
|
Placebo + MTX (N= 133)a |
SIMPONI® 50 mg + MTX* (N = 89)a |
|
|
Número de articulaciones inflamadas |
||
|
Estado basal (mediana) |
12.0 |
13.0 |
|
Semana 14 |
38% |
62% |
|
Semana 24 |
32% |
72% |
|
Número de articulaciones dolorosas |
||
|
Estado basal (mediana) |
21.0 |
26.0 |
|
Semana 14 |
30% |
60% |
|
Semana 24 |
21% |
62% |
|
Evaluación del dolor del paciente |
||
|
Estado basal (mediana) |
5.7 |
6.1 |
|
Semana 14 |
18% |
55% |
|
Semana 24 |
15% |
50% |
|
Evaluación general del paciente de la actividad de la enfermedad |
||
|
Estado basal (mediana) |
5.3 |
6.0 |
|
Semana 14 |
15% |
45% |
|
Semana 24 |
17% |
48% |
|
Evaluación general del médico de la actividad de la enfermedad |
||
|
Estado basal (mediana) |
5.7 |
6.1 |
|
Semana 14 |
35% |
55% |
|
Semana 24 |
39% |
62% |
|
Puntuación HAQ |
||
|
Estado basal (mediana) |
1.25 |
1.38 |
|
Semana 14 |
10% |
29% |
|
Semana 24 |
7% |
31% |
|
PCR (mg/dL) |
||
|
Estado basal (mediana) |
0.8 |
1.0 |
|
Semana 14 |
2% |
44% |
|
Semana 24 |
0% |
39% |
|
* p ≤ 0.001 para todas las comparaciones a N refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar en el punto cronológico. Número de articulaciones inflamadas: Se realizó el recuento de articulaciones inflamadas (0-66). Número de articulaciones dolorosas: Se realizó el recuento del articulaciones dolorosas (0-68). Evaluación de dolor del paciente: A los pacientes se les solictó que evaluaran su dolor promedio durante la semana previa en una escala VAS. El rango de la escala fue de 0 (no dolor) a 10 (peor dolor posible) cm. Evaluación general del paciente de la actividad de la enfermedad: Los pacientes evaluaron la actividad de la enfermedad en una escala VAS de la evaluación general de la actividad de la enfermedad. El rango de la escala fue de 0 (muy bien) a 10 (muy mal) cm. Evaluación general del médico de la actividad de la enfermedad: Los médicos evaluaron la actividad de la enfermedad en una escala VAS de la evaluación general de la actividad de la enfermedad. El rango de la escala fue 0 (sin artritis activa) a 10 (artritis extremadamente activa) cm. HAQ: El Índice de discapacidad del Cuestionario de Evaluación de la Salud evalua el grado de dificultad en 8 áreas funcionales (vestirse, pararse, comer, caminar, higiene, alcanzar, agarrar y actividades de la vida diaria). La mejoría de las puntuaciones HAQ (rango 0-3) se calcularon de tal manera que los valores positivos indicaron mejoría (es decir, menos discapacidad) y los valores negativos indican empeoramiento. PCR: (Rango normal 0.0-0.60 mg/dL). |
||
El porcentaje de mejoría en los componentes ACR medidos (recuento de articulaciones inflamadas, recuento de articulaciones dolorosas y PCR) observado en la semana 24 se mantuvo hasta la semana 52 y la semana 104. Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares del porcentaje de mejoría en los componentes ACR medidos (recuento de articulaciones inflamadas, recuento de articulciones dolorosas y PCR) desde la semana 104 hasta la semana 256.
Estudio 2 de AR: El tratamiento con SIMPONI® en pacientes con AR activa, previamente tratados con agente(s) anti-TNF, causó una mejoría de los signos y síntomas como se demostró con la respuesta ACR 20 en la semana 14. Una proporción significativamente mayor de pacientes alcazaron una respuesta ACR 20 en el grupo de SIMPONI® combinado y en los grupos de dosis individuales en comparación con el placebo (p≤0.001 para todas las comparaciones). En el grupo de SIMPONI® 50 mg, el porcentaje de pacientes que alcanzaron una respuesta ACR 20 fue 35%, mientras que el porcentaje en el grupo del placebo fue 18% (Tabla 5). El porcentaje de pacientes que alcanzaron una respuesta ACR 20 en la semana 24 también fue significativamente mayor en los pacientes que recibieron SIMPONI® 50 mg en comparación con el placebo (31% en comparación con 16%, respectivamente).
Cuando se consideraron las respuestas ACR 20 a través del tiempo, se observó la mejoría en la primera evaluación (semana 4) después de la primera administración de SIMPONI® 50 mg + MTX y se mantuvo hasta la semana 24 (Figura 2, Tabla 5).
Figura 2: Estudio 2 de RA: Porcentaje de pacientes que alcanzaron respuesta ACR 20 hasta la semana 24; pacientes aleatorizados en grupos de dosis del placebo y de SIMPONI® 50 mg
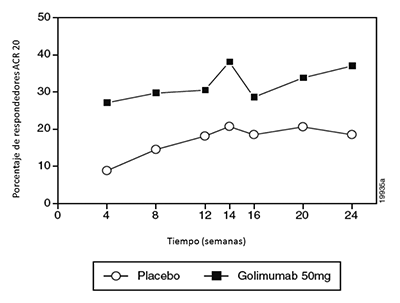
El porcentaje de los pacientes que alcanzaron respuestas ACR 50 y ACR 70 también fue significativamente mayor en el grupo de SIMPONI® 50 mg que en el grupo del placebo (Tabla 5). El porcentaje de los pacientes que alcanzaron una respuesta ACR 50 en el grupo de SIMPONI® 50 mg y del placebo fue 15% y 7%, respectivamente en la semana 14, y 16% y 4%, respectivamente en la semana 24. El porcentaje de los pacientes que alcanzaron una respuesta ACR 70 en el grupo de SIMPONI® 50 mg y del placebo fue 10% y 2%, respectivamente en la semana 14, y 9% y 2%, respectivamente en la semana 24. Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de respuestas ACR 20, ACR 50 o ACR 70 desde la semana 24 hasta la semana 256.
El porcentaje de los pacientes que alcanzaron una respuesta DAS28 (usando PCR) fue significativamente mayor para aquellos pacientes tratados con SIMPONI® 50 mg en comparación con los que recibieron placebo en la semana 14 (56% en comparación con 27%; p<0.001) y en la semana 24 (45% en comparación con 21%; p<0.001). El porcentaje de los pacientes que alcanzaron remisión DAS28 (usando PCR) fue significativamente mayor para aquellos pacientes tratados con SIMPONI® 50 mg en comparación con aquellos que recibieron el placebo en la semana 14 (9% en comparación con 1%; p<0.001) y en la semana 24 (10% en comparación con 2%; p=0.005). Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de respuesta o remisión DAS28 (usando PCR) desde la semana 24 hasta la semana 256. De igual manera, se observaron resultados estadísticamente significativos cuando se evaluó la respuesta y la remisión DAS28 (usando ESR).
|
Tabla 5: Estudio 2 de AR: Porcentaje de pacientes con AR con respuesta ACR y respuesta y remisión DAS en la semana 14 y la semana 24 |
|||
|
Placebo (N = 150)a |
SIMPONI® 50 mg (N = 147)a |
Valor-p |
|
|
ACR 20 (% de respondedores) |
|||
|
Semana 14 |
18% |
35% |
0.001 |
|
Semana 24 |
16% |
31% |
0.002 |
|
ACR 50 (% de respondedores) |
|||
|
Semana 14 |
7% |
15% |
0.021 |
|
Semana 24 |
4% |
16% |
< 0.001 |
|
ACR 70 (% de respondedores) |
|||
|
Semana 14 |
2% |
10% |
0.005 |
|
Semana 24 |
2% |
9% |
0.009 |
|
Respondedores DAS28 (usando PCR) (% de respondedores) |
|||
|
Semana 14 |
27% |
56% |
<0.001 |
|
Semana 24 |
21% |
45% |
<0.001 |
|
Remisión DAS28 (usando PCR) (% en remitentes) |
|||
|
Semana 14 |
1% |
9% |
<0.001 |
|
Semana 24 |
2% |
10% |
0.005 |
|
Respondedores DAS28 (usando ESR) (% de respondedores) |
|||
|
Semana 14 |
27% |
48% |
< 0.001 |
|
Semana 24 |
23% |
44% |
< 0.001 |
|
Remisión DAS28 (usando ESR) (% en remitentes) |
|||
|
Semana 14 |
1% |
8% |
0.003 |
|
Semana 24 |
2% |
8% |
0.015 |
|
a N refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación que puede variar en el punto cronológico. |
|||
El porcentaje de los pacientes que alcanzaron una respuesta ACR 20 fue mayor en los pacientes que recibieron SIMPONI® 50 mg que en los pacientes que recibieron placebo independientemente de la razón reportada para la descontinuación de una o más terapias anti-TNF previas. De los pacientes que reportaron la descontinuación de una o más terapias anti-TNF previas, debido a la falta de eficacia, el 35% de los que pertenecían al grupo de SIMPONI® 50 mg y el 18% de los que pertenecian al grupo del placebo alcanzaron un ACR 20 en la semana 14, mientras que en la semana 24 el porcentaje fue 29% en comparación con 16%, respectivamente. De los pacientes que reportaron la descontinuación de una o más terapias anti-TNF previas, debido a la intolerancia, el 32% de los que pertenecian al grupo de SIMPONI® 50 mg y el 17% de los que pertenecian al grupo del placebo alcanzaron un ACR 20 en la semana 14, mientras que en la semana 24 el porcentaje fue 37% en comparación con 25%, respectivamente. De los pacientes que citaron una razón diferente a la seguridad o la eficacia como razón para la descontinuación de una o más terapias anti-TNF previas, el 38% de los que pertenecian al grupo de SIMPONI® 50 mg y el 23% de los que pertenecian al grupo del placebo alcanzaron una respuesta ACR 20 en la semana 14 y en la semana 24, el porcentaje fue 35% en comparación con 21%, respectivamente (Tabla 6).
|
Tabla 6: Estudio 2 de AR: Porcentaje de respondedores ACR 20 en pacientes que reportaron descontinuación de una o más terapias anti-TNF previasa |
|||
|
Placebo |
SIMPONI® 50 mg |
Valor-p |
|
|
Respondedores ACR 20 |
|||
|
Falta de eficacia (% de respondedores) |
|||
|
N |
94 |
82 |
|
|
Semana 14 |
18% |
35% |
0.009 |
|
Semana 24 |
16% |
29% |
0.035 |
|
Intolerancia (% de respondedores) |
|||
|
N |
24 |
19 |
|
|
Semana 14 |
17% |
32% |
0.234 |
|
Semana 24 |
25% |
37% |
0.376 |
|
Otros (% de respondedores) |
|||
|
N |
62 |
58 |
|
|
Semana 14 |
23% |
38% |
0.074 |
|
Semana 24 |
21% |
35% |
0.120 |
|
a Pacientes previamente tratados con adalimumab, etanercept o infliximab. Se solicitó a los pacientes dar una razón para la descontinuación de cada terapia anti-TNF previa que hubieran recibido. |
|||
El tratamiento con SIMPONI® 50 mg también causó una mejoría significativamente mayor de cada componente ACR en comparación con el tratamiento con el placebo (Tabla 7). El recuento de articulaciones inflamadas en los grupos de SIMPONI® 50 mg y del placebo mejoró en 44% y 20%, respectivamente, en la semana 14, y en 33% y 1% respectivamente en la semana 24. La mejoría en el recuento de articulaciones dolorosas fue el 34% en comparación con 6% en la semana 14, y el 29% en comparación con -7% en la semana 24, en el grupo de SIMPONI® 50 mg y del placebo, respectivamente. Las evaluaciones de los pacientes y de los médicos y las puntuaciones del HAQ también fueron significativamente mejores con SIMPONI® 50 mg en comparación con el placebo en la semanas 14 y semana 24. Con SIMPONI® 50 mg hubo un 37% de mejoría de la PCR en comparación con 0% de mejoría con el placebo en la semana 14, y 15% en comparación con 0% en la semana 24. Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares del porcentaje de mejoría en los componentes ACR medidos (recuento de articulaciones inflamadas, recuento de articulciones dolorosas y PCR) desde la semana 24 hasta la semana 256.
|
Tabla 7: Estudio 2 de AR: Porcentaje de mejoría de los componentes ACR en la semana 14 y semana 24 en pacientes aleatorizados |
||
|
Placebo (N=135)a |
SIMPONI® 50 mg* (N=140)a |
|
|
Número de articulaciones inflamadas |
||
|
Estado basal (mediana) |
14 |
15 |
|
Semana 14 |
20% |
44% |
|
Semana 24 |
1% |
33% |
|
Número de articulaciones dolorosas |
||
|
Estado basal (mediana) |
26 |
28 |
|
Semana 14 |
6% |
34% |
|
Semana 24 |
-7% |
29% |
|
Evaluación de dolor del paciente |
||
|
Estado basal (mediana) |
7.1 |
7.0 |
|
Semana 14 |
12% |
25% |
|
Semana 24 |
4% |
25% |
|
Evaluación general del paciente de la actividad de la enfermedad |
||
|
Estado basal (mediana) |
6.7 |
6.8 |
|
Semana 14 |
8% |
29% |
|
Semana 24 |
2% |
22% |
|
Evaluación general del médico de la actividad de la enfermedad |
||
|
Estado basal (mediana) |
6.3 |
6.5 |
|
Semana 14 |
12% |
38% |
|
Semana 24 |
10% |
35% |
|
Puntuación HAQ |
||
|
Estado basal (mediana) |
1.75 |
1.63 |
|
Semana 14 |
0% |
13% |
|
Semana 24 |
0% |
11% |
|
PCR (mg/dL) |
||
|
Estado basal (mediana) |
1.0 |
0.9 |
|
Semana 14 |
0% |
37% |
|
Semana 24 |
0% |
15% |
|
* p < 0.001 para todas las comparaciones excepto para la mejoría del HAQ en la semana 14 (Donde p = 0.001) y en la semana 24 (donde p = 0.003) a N refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar en el punto cronológico. |
||
Estudio 3 de AR: El tratamiento con SIMPONI® en pacientes con AR activa sin antecedente de tratamiento con MTX causó una mejoría de los signos y síntomas según lo demostrado con la respuesta ACR 50 en la semana 24, el 38% de los pacientes del grupo combinado de SIMPONI® + MTX y 29% de los pacientes del grupo del placebo + MTX alcanzaron este criterio de evaluación (p=0.053). Cuando se realizó un análisis modificado de la intención de tratar excluyendo a 3 pacientes que fueron aleatorizados pero no tratados, la diferencia en el porcentaje de pacientes que alcanzaron una respuesta ACR 50 entre el grupo combinado de SIMPONI® + MTX y del placebo + MTX alcanzó la significancia (39% en comparación con 29%, p=0.049).
Cuando se consideraron las respuestas ACR 50 a través del tiempo, se observó mejoría en la primera evaluación (semana 4) después de una única administración de SIMPONI® 50 mg + MTX y se mantuvo hasta la semana 24 (Figura 3).
Figura 3: Estudio 3 de RA: Porcentaje de pacientes que alcanzaron respuesta ACR 50 hasta la semana 24 en pacientes aleatorizados en grupos con dosis de placebo + MTX y SIMPONI® 50 mg + MTX
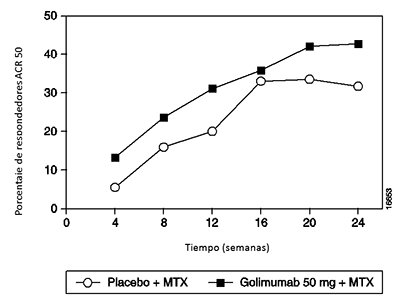
El porcentaje de los pacientes que alcanzaron respuestas ACR 20, ACR 50 y ACR 70 también fue mayor en el grupo de SIMPONI® 50 mg + MTX que en el grupo del placebo + MTX (Tabla 8). El porcentaje de pacientes que alcanzaron una respuesta ACR 20 en la semana 24 fue 62% en el grupo de SIMPONI® 50 mg + MTX en comparación con 49% en el grupo del placebo + MTX. El porcentaje de los pacientes que alcanzaron una respuesta ACR 50 en la semana 24 fue 40% en el grupo de SIMPONI® 50 mg + MTX en comparación con 29% en el grupo del placebo + MTX. El porcentaje de los pacientes que alcanzaron una respuesta ACR 70 en la semana 24 fue 24% en el grupo de SIMPONI® 50 mg + MTX y 16% en el grupo del placebo + MTX.
La proporción de los pacientes que alcanzaron una respuesta ACR 20, ACR 50 o ACR 70 se mantuvo hasta la semana 104. Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de respuestas ACR 20, ACR 50 o ACR 70 desde la semana 104 hasta la semana 256.
El porcentaje de los pacientes que alcanzaron una respuesta DAS28 (usando PCR) fue mayor en aquellos pacientes tratados con SIMPONI® 50 mg + MTX en comparación con aquellos que recibieron el placebo + MTX en la semana 24 (75% en comparación con 61%), así como lo fue el porcentaje de los pacientes que alcanzaron remisión DAS28 (usando PCR) (26% en comparación con 19%) (Tabla 8). Del mismo modo, se observaron resultados estadísticamente significativos cuando se evaluaron la respuesta y la remisión DAS28 (usando ESR).
La proporción de los pacientes que alcanzaron una respuesta DAS28 (usando PCR) en la semana 52 fue mayor en los pacientes tratados con SIMPONI® 50 mg + MTX en comparación con aquellos que recibieron el placebo + MTX (72% en comparación con 61%; p = 0.035). Del mismo modo, se observaron resultados estadísticamente significativos cuando se evaluó la respuesta DAS28 (usando ESR). El porcentaje de los pacientes que alcanzaron una remisión DAS28 (usando PCR) en la semana 52 fue mayor en los pacientes tratados con SIMPONI® 50 mg + MTX en comparación con aquellos que recibieron el placebo + MTX (35% en comparación con 23%; p=0.018). La proporción de pacientes que alcanzaron una respuesta o remisión DAS28 (usando PCR) en la semana 52 se mantuvo en la semana 104. Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se obtuvieron tasas similares de la respuesta o remisión DAS28 (con PCR) desde la semana 104 hastaa la semana 256.
El análisis de cada respuesta ACR y DAS que se muestra en la Tabla 8 usó un procedimiento de prueba jerárquico para preservar el error de Tipo 1 en 0.05. La primera prueba en este procedimiento fue estadísticamente significativa para todas las medidas con excepción de la respuesta ACR 50 (ver nota al pie de la tabla) y respuesta ACR 70.
|
Tabla 8: Estudio 3 de AR: Porcentaje de pacientes con AR con respuestas ACR y respuesta y remisión DAS en la semana 24 |
|||
|
Placebo + MTX (N = 160)a |
SIMPONI® 50 mg + MTX (N = 159)a |
Valor-p |
|
|
ACR 20 (% de respondedores) |
49% |
62% |
0.028 |
|
ACR 50 (% de respondedores) |
29% |
40% |
0.042* |
|
ACR 70 (% de respondedores) |
16% |
24% |
0.064 |
|
Respuesta DAS28 (usando PCR) (% de respondedores) |
61% |
75% |
0.007 |
|
Remisión DAS28 (usando PCR) (% de remitentes) |
19% |
26% |
0.129 |
|
Respuesta DAS28 (usando ESR) (% de respondedores) |
61% |
73% |
0.027 |
|
Remisión DAS28 (usando ESR) (%de remitentes) |
11% |
25% |
0.001 |
|
a N refleja los pacientes aleatorizados * Debido a que se consideró un enfoque jerárquico para las múltiples pruebas con el fin de preservar el error de Tipo 1 en 0.05 y el valor-p desde la primera prueba fue 0.053, para la comparación que se describió anteriormente, el valor-p no debe ser interpretado como significancia estadística. |
|||
El tratamiento con SIMPONI® 50 mg en combinación con MTX causó una mayor mejoría en cada componente ACR en comparación con el tratamiento con el placebo + MTX en la semana 24 (Tabla 9). El recuento de articulaciones inflamadas mejoró en 76% en el grupo de SIMPONI® 50 mg + MTX y 67% en el grupo del placebo + MTX. La mejoría en el recuento de articulaciones dolorosas en la semana 24 fue 67% en el grupo de SIMPONI® 50 mg + MTX y 57% en el grupo del placebo + MTX. Las evaluaciones de los pacientes y los médicos y la puntuación del HAQ también causó una mayor mejoría con SIMPONI® 50 mg + MTX en comparación con el placebo + MTX. Con SIMPONI® 50 mg + MTX, hubo una mejoría del 57% de la PCR en comparación con una mejoría del 43% con el placebo + MTX.
El porcentaje de mejoría de todos los componentes de la respuesta ACR observado en la semana 24 se mantuvo en la semana 52. La mejoría se mantuvo en los componentes ACR medidos (recuento de articulaciones inflamadas, recuento de articulaciones dolorosas y PCR) hasta la semana 104. En los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares del porcentaje de mejora de los componentes ACR medidos (recuento de articulaciones inflamadas, recuento de articulaciones dolorosas y PCR) desde la semana 104 hasta la semana 256.
El análisis de cada componente ACR que se muestra en la Tabla 9 usó un procedimiento de prueba jerárquico para preservar el error de Tipo 1 en 0.05. La primera prueba en este procedimiento fue estadísticamente significativa para todas las medidas con excepción del número de articulaciones inflamadas y la evaluación general del médico de la actividad de la enfermedad.
|
Tabla 9: Estudio 3 de AR: Porcentaje de mejoría de los componentes ACR en la semana 24 en pacientes aleatorizados |
|||
|
Placebo + MTX (N=160)a |
SIMPONI® 50 mg + MTX (N=159)a |
Valor-p |
|
|
Número de articulaciones inflamadas |
|||
|
Estado basal (mediana) |
11 |
13 |
|
|
Semana 24 |
67% |
76% |
0.127 |
|
Número de articulaciones dolorosas |
|||
|
Estado basal (mediana) |
26 |
26 |
|
|
Semana 24 |
57% |
67% |
0.023 |
|
Evaluación del dolor del paciente |
|||
|
Estado basal (mediana) |
7 |
7 |
|
|
Semana 24 |
44% |
52% |
0.028 |
|
Evaluación general del paciente de la actividad de la enfermedad |
|||
|
Estado basal (mediana) |
6 |
6 |
|
|
Semana 24 |
37% |
50% |
0.042 |
|
Evaluación general del médico de la actividad de la enfermedad |
|||
|
Estado basal (mediana) |
6 |
6 |
|
|
Semana 24 |
63% |
67% |
0.206 |
|
Puntuación HAQ |
|||
|
Estado basal (mediana) |
1.50 |
1.50 |
|
|
Semana 24 |
37% |
44% |
0.141 |
|
PCR |
|||
|
Estado basal (mediana) |
1.4 |
1.3 |
|
|
Semana 24 |
43% |
57% |
0.002 |
|
a N refleja los pacientes aleatorizados |
|||
Estudio 1 de AR por vía IV: El tratamiento con SIMPONI® IV en pacientes con AR activa a pesar del tratamiento con MTX causó una mejoría de los signos y síntomas tal como se demostró por el porcentaje de los pacientes que alcanzaron una respuesta ACR 20 en la semana 14. Un porcentaje significativamente mayor de los pacientes alcanzaron una respuesta ACR 20 en el grupo de SIMPONI® IV + MTX que en el grupo del placebo IV + MTX (p< 0.001). En el grupo de SIMPONI® IV + MTX, el 58.5% de los pacientes alcanzaron una respuesta ACR 20 en comparación con el 24.9% en el grupo del placebo IV + MTX en la semana 14. El porcentaje de los pacientes que alcanzaron una respuesta ACR 20 en la semana 24 también fue significativamente mayor en los pacientes que recibieron SIMPONI® IV + MTX en comparación con el placebo por vía IV + MTX (62.8% en comparación con 31.5%, respectivamente) (Tabla 10).
Figura 4: Estudio 1 de AR vía IV: Porcentaje de pacientes que alcanzaron respuesta ACR 20 hasta la semana 24 en pacientes aleatorizados en grupos con dosis del placebo por vía IV + MTX y SIMPONI® IV + MTX
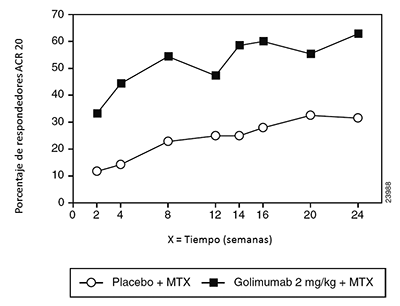
El porcentaje de los pacientes que alcanzaron respuesta ACR 50 y ACR 70 también fue mayor en el grupo de SIMPONI® IV+ MTX que en el grupo del placebo IV + MTX (Tabla 10). El porcentaje de los pacientes que alcanzaron una respuesta ACR 50 en los grupos de SIMPONI® IV+ MTX y placebo IV + MTX, fue 29.9% y 8.6%, respectivamente en la semana 14 (p<0.001) y 34.9% y 13.2%, respectivamente en la semana 24 (p<0.001). El porcentaje de los pacientes que alcanzaron una respuesta ACR 70 en los grupos de SIMPONI® IV+ MTX y placebo IV + MTX fue de 12.4% y 3.0%, respectivamente, en la semana 14 (p<0.001) y 17.5% y 4.1%, respectivamente, en la semana 24 (p<0.001).
La proporción de los pacientes que alcanzaron una respuesta ACR 20, ACR 50 o ACR 70 se mantuvo después de la semana 24 hasta la semana 52.
El porcentaje de los pacientes que alcanzaron una respuesta DAS28 (usando PCR) fue significativamente mayor en aquellos pacientes tratados con SIMPONI® IV + MTX en comparación con los que recibieron el placebo IV + MTX en la semana 14 (81.3% en comparación con 40.1%; p<0.001) y en la semana 24 (81.0% en comparación con 44.7%; p<0.001). El porcentaje de los pacientes que alcanzaron remisión DAS28 < 2.6 (usando PCR) fue significativamente mayor en aquellos pacientes tratados con SIMPONI® IV + MTX en comparación con los que recibieron el placebo IV + MTX en la semana 14 (15.4% en comparación con 4.6%; p<0.001) y en la semana 24 (17.7% en comparación con 5.1%; p<0.001). De los pacientes tratados con SIMPONI® I.V. + MTX que alcanzaron remisión DAS28, el 43% no tenía articulaciones activas, el 27% tenía una articulación activa, el 23% tenía dos articulaciones activas y el 7% tenía tres o más articulaciones activas, donde una articulación activa fue calificada como dolorosa o inflamada o ambas. La proporción de los pacientes tratados con SIMPONI® IV que alcanzaron una respuesta o remisión DAS28 (usando PCR) en la semana 24 se mantuvo hasta la semana 52.
|
Tabla 10: Estudio 1 de AR por vía IV: Porcentaje de pacientes con AR con respuesta ACR y respuestas y remisión DAS en la semana 14, semana 24 y semana 52 |
|||||||||||||||||||
|
Placebo IV + MTX (N= 197)a |
SIMPONI® IV+ MTX (N = 395)a |
Valor-p |
|||||||||||||||||
|
ACR 20 (% de respondedores) |
|||||||||||||||||||
|
Semana 14 |
24.9% |
58.5% |
p < 0.001 |
||||||||||||||||
|
Semana 24 |
31.5% |
62.8% |
p < 0.001 |
||||||||||||||||
|
Semana 52 |
N/Ab |
65.8% |
|||||||||||||||||
|
ACR 50 (% de respondedores) |
|||||||||||||||||||
|
Semana 14 |
8.6% |
29.9% |
p < 0.001 |
||||||||||||||||
|
Semana 24 |
13.2% |
34.9% |
p < 0.001 |
||||||||||||||||
|
Semana 52 |
N/Ab |
38.7% |
|||||||||||||||||
|
ACR 70 (% de respondedores) |
|||||||||||||||||||
|
Semana 14 |
3.0% |
12.4% |
p < 0.001 |
||||||||||||||||
|
Semana 24 |
4.1% |
17.5% |
p < 0.001 |
||||||||||||||||
|
Semana 52 |
N/Ab |
18.2% |
|||||||||||||||||
|
Respuesta DAS28 (usando PCR) (% de respondedores) |
|||||||||||||||||||
|
Semana 14 |
40.1% |
81.3% |
p < 0.001 |
||||||||||||||||
|
Semana 24 |
44.7% |
81.0% |
p < 0.001 |
||||||||||||||||
|
Semana 52 |
N/Ab |
81.3% |
|||||||||||||||||
|
Remisión DAS28 (usando PCR) (% en remitentes) |
|||||||||||||||||||
|
Semana 14 |
4.6% |
15.4% |
p < 0.001 |
||||||||||||||||
|
Semana 24 |
5.1% |
17.7% |
p < 0.001 |
||||||||||||||||
|
Semana 52 |
N/Ab |
20.3% |
|||||||||||||||||
|
a N refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar en el punto cronológico. b Estos resultados no fueron reportados ya que todos los pacientes recibieron SIMPONI® IV+ MTX después de la semana 24 debido al diseño del estudio. Una respuesta ACR 20 (Felson et al, 1995) se definió como: 1. ≥ 20% de mejoría en el recuento de articulaciones inflamadas (66 articulaciones) y recuento de articulaciones dolorosas (68 articulaciones); y 2. ≥ 20% de mejoría en 3 de las siguientes cinco evaluaciones: • Evaluación de dolor del paciente en una escala VAS de 0-10 cm (de ausencia de dolor al peor dolor posible). • Evaluación general del paciente de la actividad de la enfermedad en una escala VAS de 0-10 cm (de muy bien a muy mal). • Evaluación general del médico de la actividad de la enfermedad en una escala VAS de 0-10 cm (ausencia de artritis activa a artritis extremadamente activa). • Evaluación del funcionamiento físico del paciente medido por el HAQ en una escala de 0 a 3 (sin ninguna dificultad a incapacidad) • PCR. Una respuesta ACR 50 o ACR 70 se definió como ≥ 50% o ≥ 70% de mejoría en los anteriores numerales 1 y 2. La DAS28 (usando CRP) se derivó usando evaluaciones del recuento de 28 articulaciones inflamadas (inflamadas 28) y el recuento de 28 articulaciones dolorosas/sensibles (dolorosas 28), evaluación general del paciente de la actividad de la enfermedad (GH) usando la fórmula, combinada con PCR o ESR: DAS28=0.56 (raíz cuadrada(dolorosa 28)) + 0.28(raíz cuadrada(inflamada 28)) + 0.36(Ln (PCR+1)) [o 0.70)Ln(ESR)] + 0.014 (GH) + 0.96 (van der Linden, 2004; van Riel, 2000). La respuesta DAS28 en una visita se basó en la puntuación DAS28 vigente así como la mejoría desde el estado basal de la puntuación DAS28. Los pacientes se clasificaron como: Mejoría DAS28 desde el estado basal:
Los respondedores DAS28 incluyen pacientes con respuesta moderada o buena. Los remitentes DAS28 incluyen pacientes con un valor DAS28 de < 2.6 en una visita. |
|||||||||||||||||||
El tratamiento con SIMPONI® IV + MTX también causó una mejoría significativamente mayor de cada componente ACR en comparación con el tratamiento con el placebo + MTX (Tabla 11). El recuento de las articulaciones inflamadas en el grupo de SIMPONI® IV + MTX y el placebo IV + MTX mejoró en 68% y 25%, respectivamente, en la semana 14, y 75% y 27%, respectivamente, en la semana 24. La mejoría en el recuento de articulaciones dolorosas fue 62% en comparación con 13% en la semana 14 y 64% en comparación con 14% en la semana 24, en el grupo de SIMPONI® IV+ MTX y el placebo IV + MTX, respectivamente. Las evaluaciones del paciente y el médico y la puntuación del HAQ también fueron significativamente mejores con SIMPONI® IV + MTX en comparación con el placebo IV + MTX en la semanas 14 y 24. Con SIMPONI® IV+ MTX, hubo una mejoría del 78% de la PCR en comparación con una mejoría del 29% con el placebo + MTX en la semana 14 y 77% en comparación con 21% en la semana 24. La mejoría en todos los componentes ACR observada en la semana 24 se mantuvo en cada visita hasta la semana 52.
|
Tabla 11: Estudio 1 de AR vía IV: Porcentaje de mejoría de los componentes ACR en la semana 14, semana 24 y semana 52 en pacientes aleatorizados |
|||
|
Placebo IV + MTX (N= 197)a |
SIMPONI® IV+ MTX (N = 395)a |
Valor-p |
|
|
Número de articulaciones inflamadas |
|||
|
Estado basal (mediana) |
12.0 |
12.0 |
|
|
Semana 14 |
25% |
68% |
p ≤0.001 |
|
Semana 24 |
27% |
75% |
p ≤0.001 |
|
Semana 52 |
N/Ab |
86% |
|
|
Número de articulaciones dolorosas |
|||
|
Estado basal (mediana) |
22.0 |
24.0 |
|
|
Semana 14 |
13% |
62% |
p ≤0.001 |
|
Semana 24 |
14% |
64% |
p ≤0.001 |
|
Semana 52 |
N/Ab |
71% |
|
|
Evaluación del dolor del paciente |
|||
|
Estado basal (mediana) |
6.9 |
6.5 |
|
|
Semana 14 |
7% |
38% |
p ≤0.001 |
|
Semana 24 |
11% |
43% |
p ≤0.001 |
|
Semana 52 |
N/Ab |
41% |
|
|
Evaluación general del paciente de la actividad de la enfermedad |
|||
|
Estado basal (mediana) |
6.9 |
6.6 |
|
|
Semana 14 |
7% |
37% |
p ≤0.001 |
|
Semana 24 |
14% |
41% |
p ≤0.001 |
|
Semana 52 |
N/Ab |
42% |
|
|
Evaluación general del médico de la actividad de la enfermedad |
|||
|
Estado basal (mediana) |
6.3 |
6.3 |
|
|
Semana 14 |
15% |
52% |
p ≤0.001 |
|
Semana 24 |
24% |
55% |
p ≤0.001 |
|
Semana 52 |
N/Ab |
61% |
|
|
Puntuación HAQ |
|||
|
Estado basal (mediana) |
1.6 |
1.6 |
|
|
Semana 14 |
7% |
29% |
p ≤0.001 |
|
Semana 24 |
9% |
30% |
p ≤0.001 |
|
Semana 52 |
N/Ab |
30% |
|
|
PCR (mg/dL) |
|||
|
Estado basal (mediana) |
1.7 |
2.0 |
|
|
Semana 14 |
29% |
78% |
p ≤0.001 |
|
Semana 24 |
21% |
77% |
p ≤0.001 |
|
Semana 52 |
N/Ab |
66% |
|
|
a N refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar en el punto cronológico. b Estos resultados no fueron reportados ya que todos los pacientes recibieron SIMPONI® IV + MTX después de la semana 24 debido al diseño del estudio. Número de articulaciones inflamadas: Se realizó el recuento de articulaciones inflamadas (0-66). Número de articulaciones dolorosas: Se realizó el recuento del articulaciones dolorosas (0-68). Evaluación de dolor del paciente: A los pacientes se les solicitó que evaluaran su dolor promedio durante la semana previa en una escala VAS. El rango de la escala fue de 0 (no dolor) a 10 (peor dolor posible) cm. Evaluación general del paciente de la actividad de la enfermedad: Los pacientes evaluaron la actividad de la enfermedad en una escala VAS de la evaluación general de la actividad de la enfermedad. El rango de la escala fue de 0 (muy bien) a 10 (muy mal) cm. Evaluación general del médico de la actividad de la enfermedad: Los médicos evaluaron la actividad de la enfermedad en una escala VAS de la evaluación general de la actividad de la enfermedad. El rango de la escala fue 0 (sin artritis activa) a 10 (artritis extremadamente activa) cm. HAQ: El Índice de discapacidad del cuestionario de evaluación de la salud evalua el grado de dificultad en 8 áreas funcionales (vestirse, ponerse de pie, comer, caminar, higiene, alcanzar, agarrar y actividades de la vida diaria). La mejoría de las puntuaciones del HAQ (rango 0-3) se calcularon de tal manera que los valores positivos indicaron mejoría (es decir, menos discapacidad) y los valores negativos indican empeoramiento. PCR: (Rango normal 0.0-0.60 mg/dL) |
|||
Respuesta clínica principal: La respuesta clínica principal, definida como el mantenimiento de una respuesta ACR 70 durante un periodo continuo de 6 meses se midió en el Estudio 3 en AR. En la semana 52, el 15% de los pacientes en el grupo de SIMPONI® 50 mg + MTX alcanzaron un respuesta clínica mayor en comparación con el 7% de los pacientes en el grupo del placebo + MTX (p = 0.018).
Respuesta radiográfica: La progresión de daño articular estructural (erosiones y estrechamiento del espacio articular) en las manos y los pies se evaluó en el Estudio 3 en AR en la semana 52 como un criterio de evaluación co-principal y en el Estudio 1 en AR en la semana 24 como un principal criterio de evaluación secundaria. El cambio desde el estado basal de la puntuación vdH-S, una puntuación compuesta del daño estructural que mide radiograficamente el número y tamaño de las erosiones articulares y el grado de estrechamiento del espacio articular en manos/muñecas y pies se usó para evaluar el grado de daño estructural.
Estudio 3 de AR: SIMPONI® 50 mg + MTX causó una progresión radiográfica significativamente menor que el placebo + MTX, según se evaluó por la puntuación total vdH-S (p = 0.015). Los resultados se muestran en la Tabla 12.
|
Tabla 12: Estudio 3 de AR: Cambio radiográfico desde el estado basal (semana 52) |
||
|
Placebo + MTX (N=160)a |
SIMPONI® 50 mg + MTX (N=159)a |
|
|
Puntuación total |
||
|
Estado basal |
19.7 (35.4) |
18.7 (32.4) |
|
Cambio desde el estado basal |
1.4 (4.6) |
0.7 (5.2)* |
|
Puntuación de la erosión |
||
|
Estado basal |
11.3 (18.6) |
10.8 (17.4) |
|
Cambio desde el estado basal |
0.74 (2.8) |
0.48 (2.1) |
|
Puntuación del estrechamiento del espacio articular (JSN) |
||
|
Estado basal |
8.4 (17.8) |
7.9 (16.1) |
|
Cambio desde estado basal |
0.6 (2.3) |
0.2 (2.0)* |
|
a N refleja los pacientes aleatorizados. * p < 0.05. Los valores son medias [desviación estándar(SD)] de la puntuación total vdH-S. |
||
SIMPONI® 50 mg + MTX demostró una significativa inhibición de la progresión radiográfica en comparación con el placebo + MTX entre los pacientes con cambio anormal de la PCR (> 1.0 mg/dL) (media (SD) desde el estado basal en la puntuación total vdH-S de 1.3 (7.0) frente a 2.2 (5.6) respectivamente, p = 0.010). Un mayor número de pacientes en el grupo de SIMPONI® 50 mg + MTX (71%) no tuvo nuevas erosiones en articulaciones no afectadas en el estado basal en comparación con MTX solo (54%) (Tabla 13). Hubo un número significativamente mayor de pacientes en el grupo de SIMPONI® 50 mg + MTX que no presentó incremento desde el estado basal en la puntuación total vdH-S en comparación con el grupo del placebo + MTX (Tabla 14).
|
Tabla 13: Estudio 3 de AR: Nuevas erosiones en articulaciones previamente no afectadas (semana 52) |
|||
|
Placebo + MTX (N=160)a |
SIMPONI® 50 mg + MTX (N=159)a |
Valor- p |
|
|
Pacientes por lo menos con 1 articulación previamente no afectada |
141 |
140 |
|
|
Pacientes sin nuevas erosiones |
76 (54%) |
100 (71%) |
0.003 |
|
a N refleja los pacientes aleatorizados. Los valores son números (%). |
|||
|
Tabla 14: Estudio 3 de AR: Número de pacientes con cambio desde el estado basal en la puntuación total vdH-S ≤ 0 (semana 52) |
|||
|
Placebo + MTX (N=141) |
SIMPONI® 50 mg + MTX (N=140) |
Valor-p |
|
|
Pacientes con cambio desde el estado basal en la puntuación total vdH-S ≤ 0 |
76 (54%) |
100 (71%) |
0.003 |
|
Los valores son números (%) |
|||
La inhibición de la progresión radiográfica observada en la semana 52 se mantuvo en la semana 104. Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, la inhibición de la progresión radiográfica fue similar desde la semana 104 hasta la semana 256.
Estudio 1 de AR: El cambio desde el estado basal de la puntuación total vdH-S en la semana 24 en todos los grupos de tratamiento fue mínimo. No se observó diferencia significativa en el cambio desde el estado basal de la puntuación total vdH-S en la semana 24 en el grupos de SIMPONI® + MTX en comparación con el grupo del placebo + MTX.
Estudio 1 de AR por vía IV: En el Estudio 1 en AR por vía IV, el daño articular estructural se evaluó radiográficamente y se expresó como un cambio de la puntuación vdH-S en la semana 24 en comparación con el estado basal. El grupo de tratamiento con SIMPONI® IV + MTX inhibió significativamente la progresión del daño estructural en comparación con el placebo + MTX, según evaluado por la puntuación total vdH-S tal como se muestra en la Tabla 15. La inhibición de la progresión radiográfica continuó observándose en pacientes que recibieron SIMPONI® IV + MTX en la semana 52.
|
Tabla 15. Estudio 1 de AR por vía IV: Cambio radiográfico desde el estado basal en la semana 24 y en la semana 52 |
||
|
Placebo + MTX (N=197)a |
SIMPONI® IV + MTX (N=395)a |
|
|
Media (SD) |
Media (SD) |
|
|
Puntuación total vdH-S |
||
|
Estado basal |
50.3 (±59.8) |
47.6 (±54.6) |
|
Cambio desde el estado basal hasta la semana 24 |
1.1 (±3.2) |
0.03 (±1.9)* |
|
Cambio desde el estado basal hasta la semana 52 |
1.2 (±4.0)b |
0.13 (±3.1) |
|
Puntuación de la erosión |
||
|
Estado basal |
25.6 (±32.3) |
23.9 (±29.0) |
|
Cambio desde el estado basal hasta la semana 24 |
0.5 (±2.1) |
-0.1 (±1.1)* |
|
Cambio desde el estado basal hasta la semana 52 |
0.4 (±2.4)b |
-0.2 (±1.8) |
|
Puntuación JSN |
||
|
Estado basal |
24.6 (±29.5) |
23.7 (±28.3) |
|
Cambio desde el estado basal hasta la semana 24 |
0.6 (±1.7) |
0.1 (±1.3)* |
|
Cambio desde el estado basal hasta la semana 52 |
0.8 (±2.2)b |
0.3 (±2.0) |
|
a N refleja los pacientes aleatorizados. b Estos resultados se basan en pacientes que recibieron SIMPONI® IV + MTX por lo menos durante seis meses. * p ≤ 0.002. |
||
En la semana 24, una proporción significativamente mayor de pacientes en el grupo de SIMPONI® IV + MTX (71%) no presentó progresión del daño estructural (cambio de la puntuación total vdH-S ≤ 0), en comparación con el 57% de los pacientes en el grupo del placebo + MTX (Tabla 16).
|
Tabla 16: Estudio 1 de AR por vía IV: Número de pacientes con cambio desde el estado basal de la puntuación total vdH-S ≤ 0 (Semanas 24 y 52) |
|||
|
Placebo + MTX (N=197) |
SIMPONI® IV + MTX (N=395) |
Valor-p |
|
|
Pacientes con cambio desde el estado basal de la puntuación total en al escala vdH-S ≤ 0 en la semana 24 |
113 (57%) |
279 (71%) |
0.001 |
|
Pacientes con cambio desde el estado basal de la puntuación total vdH-S ≤ 0 en la semana 52 |
117 (59%)a |
278 (70%) |
|
|
a Estos resultados se basan en pacientes que recibieron SIMPONI® IV + MTX por lo menos durante seis meses. Los valores son números (%) |
|||
Mejoría del funcionamiento físico y la calidad de vida relacionada con la salud:
Estudio 1 de AR: En el Estudio 1 en AR, el funcionamiento físico y la calidad de vida relacionada con la salud se evaluaron usando el Índice de discapacidad del HAQ, el cuestionario de salud SF-36, la escala de fatiga para la Evaluación Funcional en el Tratamiento de Enfermedades Crónicas (FACIT-F) y el Cuestionario Breve del Dolor (BPI). Los pacientes tratados con SIMPONI® 50 mg + MTX mostraron una mejoría significativamente mayor del HAQ en comparación con el placebo + MTX en la semana 14 (mediana 0.38 frente a 0.13; p<0.001) y en la semana 24 (mediana 0.38 frente a 0.13; p <0.001). La media (± SD) de la mejoría del HAQ desde el estado basal hasta la semana 14 fue de 0.42 ± 0.50 en el grupo de SIMPONI® 50 mg + MTX y 0.16 ± 0.49 en el grupo del placebo + MTX. La media de la mejoría desde el estado basal hasta la semana 24 fue de 0.47 ± 0.55 en el grupo de SIMPONI® 50 mg + MTX y 0.13 ± 0.58 en el grupo del placebo (ver Tabla 17). La proporción de los pacientes que alcanzaron una mejoría clínicamente signficativa ≥ 0.25 del HAQ desde el estado basal hasta la semana 24 fue mayor en el grupo de SIMPONI® 50 mg + MTX que en el grupo del placebo + MTX (68% en comparación con 39%; no se calculó el valor-p). El ochenta y siete por ciento de los pacientes que alcanzaron una mejoría clínicamente signficativa ≥ 0.25 del HAQ desde el estado basal hasta la semana 24 en el grupo de SIMPONI® 50 mg + MTX mantuvo esta mejoría del HAQ hasta la semana 104. La proporción de los pacientes que alcanzaron una mejoría clínicamente significativa ≥ 0.3 desde el estado basal hasta la semana 24 fue mayor en el grupo de SIMPONI® 50 mg + MTX que en el grupo del placebo + MTX (53% en comparación con 34%; no se calculó el valor-p). El ochenta y cinco por ciento de los pacientes que alcanzaron una mejoría ≥ 0.3 del HAQ desde el estado basal hasta la semana 24 en el grupo de SIMPONI® 50 mg + MTX mantuvo esta mejoría del HAQ hasta la semana 104. Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de mejoría del HAQ desde la semana 104 hasta la semana 256.
|
Tabla 17: Estudio 1 de AR: Mejoría del funcionamiento físico medida por el HAQ |
||
|
Placebo + MTX (N=133)a |
SIMPONI® 50 mg + MTX* (N=89)a |
|
|
Puntuación HAQ en el estado basal |
||
|
Media ± SD |
1.32 ± 0.70 |
1.41 ± 0.691 |
|
Mediana |
1.25 |
1.38 |
|
Mejoría del HAQ |
||
|
Semana 14 |
||
|
Media ± SD |
0.16 ± 0.49 |
0.42 ± 0.50 |
|
Mediana |
0.13 |
0.38 |
|
Semana 24 |
||
|
Media ± SD |
0.13 ± 0.58 |
0.47 ± 0.55 |
|
Mediana |
0.13 |
0.38 |
|
*p < 0.001 para todas las comparaciones; los cálculos del valor-p se basan en comparaciones de los valores de las medianas. aN refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar en el punto cronológico. Las mejorías de las puntuaciones HAQ (rango 0-3) se calcularon de tal forma que los valores positivos indican mejoría (es decir, menos discapacidad) y los valores negativos indican empeoramiento. |
||
En el Estudio 1 de AR, los pacientes tratados con SIMPONI® 50 mg + MTX mostraron una mejoría significativamente mayor desde el estado basal en la puntuación del resumen del componente físico (PCS) del SF-36 en comparación con el placebo + MTX en la semana 14 (7.7 frente a 1.9; p<0.001) y en la semana 24 (8.1 frente a 2.4; p<0.001). La media de la puntuación del PCS del SF-36 en el estado basal de 30.45 ± 8.37 en el grupo de SIMPONI® 50 mg + MTX y 31.6 ± 8.3 en el grupo del placebo + MTX fueron más bajas que la normal de 50 ± 10 medida en la población de Estados Unidos. La media del cambio desde el estado basal en la puntuación del PCS del SF-36 en la semana 14 fue mayor en el grupo de SIMPONI® 50 mg + MTX que en el grupo del placebo + MTX (8.0 ± 7.2 frente a 2.4 ± 7.8, respectivamente). La media de la mejoría de la puntuación del PCS del SF-36 se mantuvo hasta la semana 24 (Tabla 18).
|
Tabla 18: Estudio 1 de AR: Mejoría de la calidad de vida relacionada con la salud medida por SF-36 |
|||
|
Placebo + MTX (N=133)a |
SIMPONI® 50 mg + MTX (N=89)a |
Valor-p* |
|
|
Puntuación del PCS del SF-36 en el estado basal |
|||
|
Media ± SD |
31.6 ± 8.3 |
30.5 ± 8.4 |
|
|
Mediana |
31.5 |
28.6 |
|
|
Mejoría del PCS del SF-36 |
|||
|
Semana 14 |
|||
|
Media ± SD |
2.4 ± 7.8 |
8.0 ± 7.2 |
|
|
Mediana |
1.9 |
7.7 |
<0.001 |
|
Semana 24 |
|||
|
Media ± DE |
2.5 ± 8.1 |
8.3 ± 8.3 |
|
|
Mediana |
2.4 |
8.1 |
<0.001 |
|
*Los cálculos del valor-p se basan en comparaciones de los valores de las medianas aN refleja los pacientes aleatorizados; el número real de pacientes evaluables para este criterio de evaluación puede variar en el punto cronológico. SF-36: El SF-36 consta de 8 escalas de múltiples preguntas: Las limitaciones del funcionamiento físico, rol física, dolor corporal, salud general, vitalidad, funcionamiento social, rol emocional y salud mental. Cuatro escalas contribuyen a la puntuación del Resumen del componente físico (PCS): Funcionamiento físico, rol físico, dolor corporal y salud general. Cuatro escalas contribuyen a la puntuación del Resumen del Componente Mental (MCS): Funcionamiento social, vitalidad, rol emocional y salud mental. |
|||
La mejoría de la puntuación del PCS del SF-36 observada en la semana 24 se mantuvo hasta la semana 52 y la semana 104. Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de mejoría de la puntuación del PCS del SF-36 desde la semana 104 hasta la semana 256.
La mediana del cambio desde el estado basal de la puntuación FACIT-F fue significativamente mayor en el grupo de SIMPONI® 50 mg + MTX en comparación con el grupo del placebo + MTX en la semana 14 (7.0 frente a 2.0; p < 0.001) y en la semana 24 (7.0 frente a 3.0; p < 0.001). La media del cambio desde el estado basal de las puntuaciones FACIT-F fue mayor en el grupo de SIMPONI® 50 mg + MTX que en el grupo del placebo + MTX en la semana 14 (7.58 ± 8.93 frente a 2.27 ± 9.24, respectivamente). La media de la mejoría de la puntuación FACIT-F se mantuvo hasta la semana 24 en el grupo de SIMPONI® 50 mg + MTX en comparación con el placebo (7.30 ± 8.65 frente a 2.16 ± 9.53 respectivamente; Tabla 19). Se observó una mejoría significativa de la mediana del cambio desde el estado basal en el cuestionario breve del dolor (BPI) en el grupo de SIMPONI® 50 mg + MTX en comparación con el placebo en la semana 14 (–2.0 frente a –0.3; p<0.001).
|
Tabla 19: Estudio 1 de AR: Mejoría de la calidad de vida relacionada con la salud medida en la escala FACIT-F y por el BPI |
||
|
Placebo + MTX (N=133)a |
SIMPONI® 50 mg + MTX (N=89)a, * |
|
|
Puntuación FACIT-F en el estado basal |
||
|
Media ± SD |
28.65 ± 10.46 |
26.55 ± 10.97 |
|
Mediana |
28.00 |
26.00 |
|
Mejoría en FACIT-F |
||
|
Semana 14 |
||
|
Media ± SD |
2.27 ± 9.24 |
7.58 ± 8.93 |
|
Mediana |
2.0 |
7.0 |
|
Semana 24 |
||
|
Media ± SD |
2.16 ± 9.53 |
7.30 ± 8.65 |
|
Mediana |
3.0 |
7.0 |
|
Puntuación BPI en el estado basal |
||
|
Media ± SD |
4.78 ± 1.96 |
4.83 ± 1.81 |
|
Mediana |
5.0 |
5.0 |
|
Mejoría en BPI |
||
|
Semana 14 |
||
|
Media ± SD |
-0.55 ± 1.87 |
-1.88 ± 2.12 |
|
Mediana |
-0.3 |
-2.0 |
|
*p < 0.001 para todas las comparaciones; los cálculos del valor-p se basan en comparaciones de los valores de las medianas. aN refleja los pacientes aleatorizados; el número real de los pacientes evaluables para cada criterio de evaluación puede variar en el tiempo cronológico. FACIT-F – La escala de fatiga para la evaluación funcional en el tratamiento de enfermedades crónicas (FACIT-F) es un cuestionario que evalua el autoreporte de cansancio, debilidad y dificultad para llevar a cabo actividades usuales debido a la fatiga. La puntuación total FACIT-F varía de 0 a 52, donde una puntuación más alta indica menos fatiga. BPI - El cuestionario breve del dolor (BPI) es un cuestionario autoadministrado desarrollado para evaluar el dolor en la investigación clínica. La escala es de 0-10, donde las puntuaciones más bajas indican dolor más leve. |
||
Estudio 2 de AR: En el Estudio 2 en AR, se evaluó el funcionamiento físico y la calidad de vida relacionada con la salud usando el índice de discapacidad del HAQ y la puntuación FACIT-F. Los pacientes tratados con SIMPONI® 50 mg mostraron una media significativamente mayor de la mejoría del HAQ en comparación con el placebo en la semana 24 (0.13 frente a 0.00; p <0.001). La media (± SD) de la mejoría del HAQ desde el estado basal hasta la semana 24 fue de 0.23 ± 0.50 en el grupo tratado con SIMPONI® y de 0.03 ± 0.50 en el grupo del placebo (ver Tabla 20). Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de mejoría del HAQ desde la semana 24 hasa la semana 256.
|
Tabla 20: Estudio 2 de AR: Mejoría del funcionamiento físico medida por el HAQ |
||
|
Placebo N=150a |
SIMPONI® 50 mg* N=147a |
|
|
Puntuación HAQ en el estado basal |
||
|
Media ± SD |
1.63 ± 0.63 |
1.58 ± 0.65 |
|
Mediana |
1.75 |
1.63 |
|
Mejoria del HAQ |
||
|
Semana 24 |
||
|
Media ± SD |
0.03 ± 0.50 |
0.23 ± 0.50 |
|
Mediana |
0.00 |
0.13 |
|
* p < 0.001; los cálculos del valor-p se basan en comparaciones de los valores de las medianas. a N refleja los pacientes aleatorizados. La mejoría de la puntuación del HAQ (rango 0-3) se calculó de tal manera que los valores positivos indican mejoría (es decir, menos discapacidad) y los valores negativos indican empeoramiento. |
||
La mediana del cambio desde el estado basal de la puntuación FACIT-F en la semana 14 fue significativamente mayor en el grupo de SIMPONI® 50 mg en comparación con el placebo (6.0 frente a 1.0; p=0.001). La media (± SD) del cambio desde el estado basal de la puntuación FACIT-F en la semana 14 fue de 6.02 ± 10.14 en el grupo de SIMPONI® 50 mg en comparación con 2.16 ± 9.74 en el grupo del placebo. La mediana de la mejoría desde el estado basal observada en la semana 14 en el grupo de SIMPONI® 50 mg se sostuvo en la semana 24 y permaneció estadísticamente superior al placebo (p = 0.043) (ver Tabla 21).
|
Tabla 21: Estudio 2 de AR: Mejoría de la fatiga medida en la escala FACIT-F en pacientes aleatorizados |
||
|
Placebo N=150a |
SIMPONI® 50 mg* N=147a |
|
|
Puntuación FACIT-F en el estado basal |
||
|
Media ± SD |
23.74 ± 10.34 |
23.07 ± 12.31 |
|
Mediana |
22.0 |
23.0 |
|
Mejoría de la puntuación FACIT-F |
||
|
Semana 14 |
||
|
Media ± SD |
2.16 ± 9.74 |
6.02 ± 10.14 |
|
Mediana |
1.0 |
6.0 |
|
Semana 24 |
||
|
Media ± SD |
2.83 ± 9.74 |
5.37 ± 11.79 |
|
Mediana |
3.0 |
5.0 |
|
* p = 0.001 en la semana 14 y p= 0.043 en la semana 24; los cálculos del valor-p se basan en comparaciones de los valores de las medianas. a N refleja los pacientes aleatorizados; el número real de pacientes evaluables para este criterio de evaluación puede variar en el punto cronológico. |
||
Estudio 1 de AR por vía IV: En el Estudio 1 en AR vía IV, el funcionamiento físico y la calidad de vida relacionada con la salud se evaluaron usando el índice de discapacidad del HAQ, del SF-36 y la escala de fatiga para la Evaluación Funcional en el Tratamiento de Enfermedades Crónicas (FACIT-F). Los pacientes tratados con SIMPONI® IV + MTX mostraron medianas significativamente mayores de la mejoría del HAQ en comparación con el placebo IV + MTX en la semana 14 (mediana 0.50 frente a 0.12; p<0.001) y en la semana 24 (mediana 0.50 frente a 0.12; p <0.001). La media (± SD) de la mejoría del HAQ desde el estado basal hasta la semana 14 fue de 0.50 ± 0.58 en el grupo de SIMPONI® IV + MTX y 0.19 ± 0.56 en el grupo del placebo IV + MTX. La media de la mejoría desde el estado basal hasta la semana 24 fue 0.53 ± 0.64 en el grupo de SIMPONI® IV + MTX y de 0.20 ± 0.55 en el grupo del placebo (ver Tabla 22). La media de la mejoría del HAQ se mantuvo hasta la semana 52. La proporción de sujetos que alcanzaron una mejoría clínicamente signficativa ≥ 0.25 del HAQ desde el estado basal hasta la semana 14 fue mayor en el grupo de SIMPONI® IV + MTX que en el grupo del placebo IV + MTX (68% en comparación con 43%; p <0.001). Esta respuesta se mantuvo en la semana 24 (67% en el grupo de SIMPONI® IV + MTX frente a 45% en el grupo del placebo IV + MTX; p < 0.001) y hasta la semana 52.
|
Tabla 22: Estudio 1 de AR por vía IV: Mejoría del funcionamiento físico medida por el HAQ |
||
|
Placebo IV + MTX (N=197)a |
SIMPONI® IV + MTX* (N=395)a |
|
|
Puntuación HAQ en el estado basal |
||
|
Media ± SD |
1.57 ± 0.62 |
1.56 ± 0.67 |
|
Mediana |
1.63 |
1.63 |
|
Mejoría del HAQ |
||
|
Semana 14 |
||
|
Media ± SD |
0.19 ± 0.56 |
0.50 ± 0.58 |
|
Mediana |
0.12 |
0.50 |
|
Semana 24 |
||
|
Media ± SD |
0.20 ± 0.55 |
0.53 ± 0.64 |
|
Mediana |
0.12 |
0.50 |
|
Semana 52 |
||
|
Media ± SD |
N/Ab |
0.51 ± 0.65 |
|
Mediana |
N/Ab |
0.38 |
|
*p < 0.001 para todas las comparaciones; los cálculos del valor-p se basan en comparaciones de los valores de las medianas. aN refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar en el punto cronológico. La mejoría de la puntuación del HAQ (rango 0-3) se calculó de tal manera que los valores positivos indican mejoría (es decir, menos discapacidad) y los valores negativos indican empeoramiento. bEstos resultados no se reportaron ya que todos los pacientes recibieron SIMPONI® IV + MTX después de la semana 24 debido al diseño del estudio. |
||
En el Estudio 1 en AR por vía IV, la mediana de los cambios desde el estado basal de la puntuación del resumen del componente físico del SF-36 y de la puntuación del resumen del componente mental fueron significativamente mayores en los pacientes tratados con SIMPONI® IV + MTX en comparación con los pacientes tratados con placebo IV + MTX en la semanas 12, semana 16 y semana 24 (p<0.001 por cada componente en todos los puntos de evaluación). La mejoría de la puntuación del componente físico del SF-36 y de la puntuación del componente mental se mantuvo hasta la semana 52.
SIMPONI® IV + MTX mostró una mediana de la mejoría significativamente mayor desde el estado basal de la puntuación del resumen del componente físico (PCS) del SF-36 en comparación con el placebo IV + MTX en la semana 12 (5.6 frente a 2.6; p<0.001), en la semana 16 (7.1 frente a 1.9; p<0.001) y en la semana 24 (7.4 frente a 3.3; p<0.001). En el estado basal, la media de la puntuación del PCS del SF-36 de 30.8 ± 6.8 en el grupo de SIMPONI® I.V. + MTX y 30.9 ± 7.3 en el grupo del placebo IV + MTX fueron más bajas que la normal de 50 ± 10 medida en la población de Estados Unidos. La media de la mejoría desde el estado basal de la puntuación del PCS del SF-36 en la semana 12 fue mayor en el grupo de SIMPONI® IV + MTX que en el grupo del placebo IV + MTX (5.9 ± 7.7 frente a 3.2 ± 7.4, respectivamente). La media de la mejoría de la puntuación del PCS del SF-36 se mantuvo en la semana 16, 24 y 52.
SIMPONI® IV + MTX mostró una mediana de la mejoría significativamente mayor desde el estado basal de la puntuación del resumen del componente mental (MCS) del SF-36 en comparación con el placebo IV + MTX en la semana 12 (4.3 frente a 0.6; p<0.001), en la semana 16 (6.4 frente a 1.9; p<0.001) y en la semana 24 (6.4 frente a 1.1; p<0.001). La media del cambio desde el estado basal de la puntuación del MCS del SF-36 en la semana 12 fue mayor en el grupo de SIMPONI® IV + MTX que en el grupo del placebo IV + MTX (4.9 ± 10.3 frente a 1.5 ± 9.9, respectivamente). La media de la mejoría de la puntuación del MCS en el SF-36 se mantuvo en la semana 16, 24 y 52.
Además, las ocho escalas del SF-36 que comprende la puntuación del PCS y del MCS mostraron una mayor mediana de la mejoría en la semanas 12, semana 16 y semana 24 en el grupo de SIMPONI® IV + MTX en comparación con el grupo del placebo IV + MTX (p < 0.001 para todas las semanas y todas las escalas). En comparación con la semana 24, la mejoría en cada una de las ocho escalas del SF-36 se mantuvo hasta la semana 52.
Los pacientes tratados con SIMPONI® IV + MTX tuvieron una mediana clínicamente significativa de la mejoría de la fatiga medida en la escala FACIT-F. La mediana del cambio desde el estado basal de la puntuación en la escala FACIT-F fue significativamente mayor con SIMPONI® IV + MTX que con el placebo IV + MTX en la semana 16 (7.0 frente a 2.0; p < 0.001) y en la semana 24 (8.0 frente a 2.0; p < 0.001). La mejoría en la escala FACIT-F observada en la semana 24 se mantuvo hasta la semana 52. La mediana del cambio desde el estado basal de la escala FACIT-F hasta las semana 16 y semana 24 en el grupo de SIMPONI® IV + MTX fue de ≥ 4 puntos, lo cual indicó una mejoría clínicamente significativa de la fatiga. La media del cambio desde el estado basal de la puntuacioón en la escala FACIT-F fue mayor en el grupo de SIMPONI® IV + MTX que en el grupo del placebo IV + MTX en la semana 16 (7.54 ± 10.546 frente a 2.16 ± 9.700, respectivamente). La media de la mejoría de la puntuación en la escala FACIT-F se mantuvo hasta la semana 24 en el grupo de SIMPONI® IV + MTX en comparación con el placebo (7.96 ± 10.793 frente a 2.54 ± 10.219 respectivamente, p<0.001). Además, la proporción de los pacientes que alcanzaron una mejoría clínicamente significativa (≥ 4 puntos) de la escala FACIT-F en la semana 24 se mantuvo hasta la semana 52.
Economía sanitaria: Los datos de economía sanitaria sobre utilización de recursos sanitarios, tiempo laboral perdido por los pacientes y cuidadores, empleabilidad y productividad del paciente en el trabajo, el colegio, o el hogar se recogieron por medio de cuestionarios aplicados en el estado basal y posteriormente cada 8 semanas. Se les solicitó a los pacientes que indicaran cuánto su enfermedad afectó su productividad en las 4 semanas previas usando una escala VAS de 0 a 10 cm (de nada afectada a sumamente afectada). A continuación se presentan solamente aquellas evaluaciones en las cuales se observaron diferencias estadísticamente significativas entre SIMPONI® ± MTX y placebo ± MTX.
Estudio 1 de AR: En el estado basal, el grupo de SIMPONI® 50 mg + MTX y el grupo del placebo + MTX tuvieron una puntuación comparable de la mediana de la productividad (6.2 frente a 5.8, respectivamente). La mediana de la mejoría autoreportadas sobre la productividad fue significativamente mayor en el grupo de SIMPONI® 50 mg + MTX que en el grupo del placebo + MTX en la semana 24 (-1.9 frente a -0.4; p < 0.001). La media ± SD de la mejoría autoreportadas sobre la productividad en el grupo de SIMPONI® 50 mg + MTX y en el grupo del placebo + MTX en la semana 24 fue de –1.97 ± 3.12 frente a -0.45 ± 2.98.
Estudio 2 de AR: En el estado basal, el grupo de SIMPONI® 50 mg y el grupo del placebo tuvieron una puntuación comparable de la mediana de la productividad (7.3 frente a 7.2). La mediana de la mejoría autoreportada sobre la productividad fue significativamente mayor en el grupo de SIMPONI® 50 mg que en el grupo del placebo en la semana 16 (–1.1 frente a –0.1; p< 0.001) y en la semana 24 (–1.4 frente a –0.1; p< 0.001). La media ± SD de la mejoría autoreportadas sobre la productividad en el grupo de SIMPONI® 50 mg y el grupo del placebo en la semana 16 y en la semana 24 fue de -1.42 ± 2.76 frente a -0.27 ± 2.47 y de -1.77 ± 2.90 frente a -0.52 ± 2.79, respectivamente.
Estudio 1 en AR vía IV: La mediana del cambio desde el estado basal en la escala VAS del estado de salud (EQ VAS) fue significativamente mayor en el grupo de SIMPONI® IV+ MTX que en el grupo del placebo IV + MTX en la semanas 16 y 24. En la semana 24, la mediana de la mejoría desde el estado basal en la escala EQ VAS fue de 20.00 en el grupo de SIMPONI® IV + MTX en comparación con 6.00 en el grupo del placebo + MTX (p < 0.001) y superó el umbral del cambio clínicamente significativo definido como ½ de la desviación estándar del valor en el estado basal (24.86/2=12.5).
La disminución de la mediana en el autoreporte sobre el impacto de la enfermedad en la productividad fue significativamente mayor en el grupo de SIMPONI® IV + MTX en comparación con el grupo del placebo IV + MTX en la semana 24 (–2.9 frente a –0.5; p< 0.001). La disminución de la media ± SD en el autoreporte sobre el impacto de la enfermedad en la productividad en el grupo de SIMPONI® IV + MTX y del placebo IV + MTX en la semana 24 fue de -2.78 ± 2.92 frente a -1.03 ± 2.96, respectivamente (p< 0.001).
Artritis psoriásica:
Estudio de APs: Se evaluó la seguridad y eficacia de SIMPONI® en un estudio individual en APs (GO-REVEAL), el estudio multicéntrico, aleatorizado, doble ciego y controlado por placebo (hasta la semana 24) evaluó el tratamiento con SIMPONI® 50mg o 100 mg administrado como inyección subcutánea cada 4 semanas en 405 pacientes adultos con APs activa. Todos los pacientes que recibieron el placebo recibieron SIMPONI® 50 mg después de la semana 24, pero el ensayo permaneció doble ciego hasta que todos los pacientes completaron las 52 semanas de tratamiento. En la semana 52, los pacientes ingresaron a la fase de extensión a largo plazo en el cual los pacientes continuaron el tratamiento con SIMPONI® 50 mg o SIMPONI® 100 mg. Después que el último paciente completó la visita de la semana 52 y el ensayo fue dado a conocer, a los pacientes que recibieron SIMPONI® 50 mg se les pudo incrementar la dosis a 100 mg a discreción del investigador. Los pacientes incluidos en este estudio fueron hombres y mujeres con diagnóstico de APs de por lo menos 6 meses con un grado de lesión psoriásica en la piel por lo menos de 2 cm de diámetro y enfermedad activa por lo menos con 3 articulaciones inflamadas y 3 articulaciones dolorosas a pesar de la terapia con fármacos antirreumáticos modificadores de la enfermedad (FAMES) o antiinflamatorios no esteroideos (AINEs). No se permitió tratamiento previo con un agente anti-TNFa. Los pacientes con cada subtipo de artritis psoriásica fueron incluidos, incluyendo artritis poliarticular sin nódulos reumatoides (43%), artritis periférica asimétrica (30%), artritis de las articulaciones interfalángicas distales (DIP) (15%), espondilitis con artritis periférica (11%) y artritis mutilante (1%). Los datos de la eficacia fueron recogidos y analizados en la semana 256.
Los pacientes fueron aleatorizados para recibir placebo (N=113), SIMPONI® 50 mg (N=146) y SIMPONI® 100 mg (N=146). Los criterios de evaluación co-principales fueron el porcentaje de los pacientes que alcanzaron una respuesta ACR 20 en la semana 14 y el cambio desde el estado basal de la puntuación total vdH-S modificada para APs en la semana 24. Los principales criterios de evaluación secundaria incluyeron el porcentaje de los pacientes que alcanzaron una respuesta ACR 20 en la semana 24, respuesta 75 del Índice de Severidad y Área de Psoriasis (PASI) en la semana 14 en un subconjunto de pacientes con ≥ 3% del Área de Superficie Corporal (ASC) de la piel con psoriasis en el estado basal, mejoría de la puntuación del HAQ desde el estado basal hasta la semana 24 y cambios de la puntuación del PCS del SF-36 desde el estado basal hasta la semana 14.
Estudio por vía IV de APs: La eficacia y seguridad de SIMPONI® IV se evaluaron en un estudio multicéntrico, aleatorizado, doble ciego, controlado por placebo (GO-VIBRANT) en 480 adultos con artritis psoriásica activa a pesar de la terapia con fármacos antiinflamatorios no esteroideos (AINES) o con fármacos antirreumáticos modificadores de la enfermedad (FAMES). Los pacientes en este estudio tuvieron un diagnóstico de APs con una duración de por lo menos 6 meses y presentaron síntomas de la enfermedad activa (≥ 5 articulaciones inflamadas, ≥ 5 articulaciones sensibles y un nivel de PCR de ≥ 0.6 mg/dL). Los pacientes fueron aleatorizados para recibir SIMPONI® IV 2 mg/kg (N=241) o placebo (N=239) como una infusión intravenosa de 30 minutos en la semana 0, 4, 12 y 20. Todos los pacientes con placebo recibieron SIMPONI® IV en la semana 24, en la semana 28 y posteriormente cada ocho semanas hasta la semana 52. Los pacientes en el grupo tratado con SIMPONI® IV continuaron recibiendo infusiones de SIMPONI® IV en la semana 28 y cada ocho semanas hasta la semana 52. No se permitieron tratamientos previos con un agente biológico.
Se permitió a los pacientes continuar con dosis estables de MTX, AINEs y corticosteroides orales a dosis baja (equivalente a ≤ 10 mg de prednisona por día) durante el estudio. Al momento del enrolamiento en el estudio, se prohibió el uso de otros FAMES incluyendo agentes citotóxicos u otros agentes biológicos.
Se enrolaron pacientes con cada subtipo de APs, incluyendo artritis poliarticular con ausencia de nódulos reumatoides (44%), artritis periférica asimétrica (19%), afectación de articulaciones interfalángicas distales (8.1%), espondilitis con artritis periférica (25%) y artritis mutilans (4.8%). La mediana de la duración de la enfermedad APs fue de 3.5 años, el 86% de los pacientes había utilizado previamente MTX, y el 35% de los pacientes recibido por lo menos algún otro FAME en el pasado. En el estado basal, el 76% y el 54% de los pacientes presentaron entesitis y dactilitis, respectivamente. Durante el estudio, el uso de medicamentos concomitantes fue MTX (70%), corticosteroides orales (28%) y AINEs (71%).
El criterio de evaluación principal fue el porcentaje de los pacientes que alcanzaron una respuesta ACR 20 en la semana 14. Los criterios de valoración secundarios importantes fueron el cambio en el estado basal en la puntuación HAQ-DI en la semana 14, la proporción de sujetos que alcanzaron una respuesta ACR 50 en la semana 14, la proporción de sujetos (con afectación psoriásica del área de superficie corporal [ASC] ≥3%) que alcanzaron una respuesta PASI 75 en la semana 14, y el cambio desde el estado basal en la puntuación total modificada vdH-S en la semana 24.
Reducción de los signos y síntomas:
Estudio de APs: En general, no se apreciaron diferencias clínicamente significativas en la medida de la eficacia entre los regímenes de dosificación de SIMPONI® 50 mg y 100 mg en el estudio de fase 3 en APs hasta la semana 104. Según el diseño del estudio, a los pacientes en la extensión de largo plazo se les podia cambiar las dosis a 50 mg y 100 mg de SIMPONI® a discreción del médico del estudio.
El tratamiento con SIMPONI® causó una mejoría de los signos y síntomas según lo demostrado con el porcentaje de los pacientes que alcanzaron una respuesta ACR 20 en la semana 14. Un porcentaje significativamente mayor de los pacientes alcanzaron una respuesta ACR 20 en el grupo combinado (SIMPONI® 50 mg y SIMPONI® 100 mg) y en los grupos de dosificación individual de SIMPONI® que en el grupo del placebo (p<0.001 para todas las comparaciones). En el grupo de SIMPONI® 50 mg, el 51% de los pacientes alcanzaron una respuesta ACR 20 en comparación con el 9% en el grupo del placebo (Tabla 23). La respuesta observada en los grupos tratados con SIMPONI® fue similar en los pacientes que recibieron y que no recibieron MTX concomitantemente. Además, el porcentaje de los pacientes que alcanzaron una respuesta ACR 20 en la semana 24 fue significativamente mayor en los pacientes que recibieron SIMPONI® (52% en el grupo de 50 mg) en comparación con los pacientes que recibieron el placebo (12%).
Cuando se consideraron las respuestas ACR 20 a través del tiempo, se observó la mejoría en la primera evaluación (semana 4) después de la primera administración de SIMPONI® y se mantuvo hasta la semana 24 (Figura 5, Tabla 23).
Figura 5: Porcentaje de respondedores ACR 20 con APs hasta la semana 24; pacientes aleatorizados en grupos con dosis de placebo y SIMPONI® 50 mg
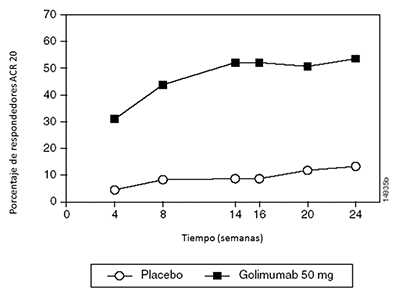
El porcentaje de los pacientes que alcanzaron respuesta ACR 50 y ARC 70 también fue mayor en los pacientes del grupo de SIMPONI® 50 mg en comparación con el grupo del placebo (Tabla 23). El porcentaje de los pacientes que alcanzaron una respuesta ACR 50 en la semana 14 fue 30% con SIMPONI® 50 mg y 2% con el placebo (p<0.001). En la semana 24, el porcentaje de los pacientes que alcanzaron una respuesta ACR 50 fue 32% y 4% (p<0.001) con SIMPONI® 50 mg y con el placebo, respectivamente. El porcentaje de los pacientes que alcanzaron una respuesta ACR 70 fue 12% y 1% en la semana 14, y 19% y 1% en la semana 24 (p<0.001) con SIMPONI® 50 mg y con el placebo, respectivamente. La proporción de los pacientes que alcanzaron respuesta ACR 20, ACR 50 o ACR 70 se mantuvieron hasta la semana 104. En los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de respuestas ACR 20, ACR 50 o ACR 70 desde la semana 104 hasta la semana 256. La respuesta observada en el grupo de SIMPONI® 50 mg fue similar en los pacientes que recibieron y que no recibieron MTX concomitantemente.
El porcentaje de los pacientes que alcanzaron el Criterio de Respuesta en Artritis Psoriásica (PsARC) y respuesta DAS28 también fue significativamente mayor en el grupo de SIMPONI® 50 mg que en el grupo del placebo en la semanas 14 y semana 24 (Tabla 23). La proporción de los pacientes que alcanzaron el PsARC o la respuesta DAS28 en la semana 24 se mantuvo hasta la semana 104. En los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI® se observaron tasas similares de respuesta en el PsARC o la respuesta DAS28 desde la semana 104 a la semana 256.
|
Tabla 23: Estudio de APs: Porcentaje de pacientes con respuesta ACR, PsARC y DAS28 en pacientes aleatorizados |
||
|
Placebo (N = 113)a |
SIMPONI® 50 mg* (N = 146)a |
|
|
ACR 20 (% de respondedores) |
||
|
Semana 14 |
9% |
51% |
|
Semana 24 |
12% |
52% |
|
ACR 50 (% de respondedores) |
||
|
Semana 14 |
2% |
30% |
|
Semana 24 |
4% |
32% |
|
ACR 70 (% de respondedores) |
||
|
Semana 14 |
1% |
12% |
|
Semana 24 |
1% |
19% |
|
PsARC (% de respondedores) |
||
|
Semana 14 |
21% |
73% |
|
Semana 24 |
29% |
70% |
|
DAS28 (% de respondedores) |
||
|
Semana 14 |
24% |
69% |
|
Semana 24 |
22% |
65% |
|
*p < 0.001 para todas las comparaciones aN refleja los pacientes aleatorizados; el número real de los pacientes evaluables para cada criterio de evaluación puede variar en el tiempo. |
||
El tratamiento con SIMPONI® también causó una mejoría significativamente mayor en comparación con el placebo de cada componente ACR (Tabla 24). El recuento de articulaciones inflamadas mejoró en 60% en comparación con 8% en la semana 14 y en 67% en comparación con 0% en la semana 24, en los grupos de SIMPONI® 50 mg y del placebo, respectivamente. En el recuento de articulaciones dolorosas, los pacientes del grupo de SIMPONI® 50 mg tuvieron una mejoría del 54% en comparación con 0% con el placebo en la semana 14 y una mejoría del 66% en comparación con el –6% en la semana 24. La evaluación de los pacientes y los médicos y la puntuación HAQ también fueron mejores con SIMPONI® 50 mg en comparación con el placebo en ambos puntos cronológicos. Con SIMPONI® 50 mg, la mejoría de la PCR fue 40% en la semana 14 y de 29% en la semana 24 en comparación con 0% de mejoría con el placebo en ambos puntos cronológicos.
|
Tabla 24: Estudio de APs: Porcentaje de mejoría de los componentes ACR en la semana 14 y 24; pacientes aleatorizados |
||
|
Placebo (N=113)a |
SIMPONI® 50 mg* (N=146)a |
|
|
Número de articulaciones inflamadas |
||
|
Estado basal (mediana) |
10.0 |
11,0 |
|
Semana 14 |
8% |
60% |
|
Semana 24 |
0% |
67% |
|
Número de articulaciones dolorosas |
||
|
Estado basal (mediana) |
18.0 |
19.0 |
|
Semana 14 |
0% |
54% |
|
Semana 24 |
-6% |
66% |
|
Evaluación del dolor del paciente |
||
|
Estado basal (mediana) |
5.4 |
5.8 |
|
Semana 14 |
-1% |
48% |
|
Semana 24 |
-2% |
50% |
|
Evaluación general del paciente de la actividad de la enfermedad |
||
|
Estado basal (mediana) |
5.2 |
5.2 |
|
Semana 14 |
2% |
49% |
|
Semana 24 |
-2% |
52% |
|
Evaluación general del médico de la actividad de la enfermedad |
||
|
Estado basal (mediana) |
5.2 |
5.4 |
|
Semana 14 |
7% |
59% |
|
Semana 24 |
5% |
71% |
|
Puntuación HAQ |
||
|
Estado basal (mediana) |
1.0 |
1.0 |
|
Semana 14 |
0% |
28% |
|
Semana 24 |
0% |
33% |
|
PCR (mg/dL) |
||
|
Estado basal (mediana) |
0.60 |
0.60 |
|
Semana 14 |
0% |
40% |
|
Semana 24 |
0% |
29% |
|
* p < 0.001 para todas las comparaciones a N refleja los pacientes aleatorizados; el número real de los pacientes evaluables para cada criterio de evaluación puede variar en el punto cronológico. PCR: Rango normal: 0.0-0.60 mg/dL |
||
El porcentaje de mejoría de los componentes ACR observado en la semana 24 se mantuvo hasta la semana 104. En los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de mejoría en los componentes ACR desde la semana 104 hasa la semana 256.
Además, los pacientes tratados con SIMPONI® 50 mg tuvieron una mejoría significativamente mayor desde el estado basal de la puntuación de entesitis medida por la puntuación Maastricht para la Entesitis en Espondilitis Anquilosante (MASES) modificada para APs en la semanas 14
y en la semana 24 (Tabla 25). La mejoría de la puntuación de la dactilitis fue numéricamente mayor en el grupo de tratamiento con SIMPONI® 50 mg comparado con el grupo de tratamiento con placebo en la semana 14 y la semana 24 (p=No significativa).
|
Tabla 25: Estudio de APs: Porcentaje de mejoría de la puntuación de la entesitis en pacientes con entesitis en el estado basal y porcentaje de mejoría de la puntuación de la dactilitis en pacientes con dactilitis en el estado basal; pacientes aleatorizados |
|||
|
Placebo (N=113) |
SIMPONI® 50 mg (N=146) |
Valor-p |
|
|
Puntuación de la entesitis |
|||
|
Pacientes con entesitis en el estado basal |
88 |
109 |
|
|
Puntuación en el estado basal (mediana) |
4.0 |
4.0 |
|
|
Porcentajes de mejoría |
|||
|
Semana 14 |
0% |
50% |
< 0.001 |
|
Semana 24 |
12% |
60% |
< 0.001 |
|
Puntuación de la dactilitis |
|||
|
Pacientes con dactilitis en el estado basal |
38 |
50 |
|
|
Puntuación en el estado basal (mediana) |
2.0 |
4.5 |
|
|
Porcentajes de mejoría |
|||
|
Semana 14 |
0% |
76% |
N.S. |
|
Semana 24 |
42% |
100% |
N.S. |
|
Entesitis: El índice de Maastricht para la puntuación de la entesitis en EA (MASES) (Heuft-Dorenbosch et al, 2003) fue desarrollado para evaluar la entésitis en pacientes con EA e incluye 13 lugares de entesis relevantes. En los pacientes con APs, el índice MASES se modificó para incluir 2 sitios adicionales (inserción izquierda y derecha de la fascia plantar en el calcáneo) de manera que se evaluó un total de 15 sitios de entesis como dolorosos (puntuación de 1) o no dolorosos (puntuación de 0) mediante la aplicación de presión local en cada uno de los sitios. La puntuación de la entesitis es la sumatoria de la puntuación de cada sitio de entesis y puede variar de 0 a 15. Se considera que un paciente tiene entésitis si la puntuación es > 0. Dactilitis: La dactilitis es una inflamación dolorosa de todo el dedo causada por inflamación de las articulaciones del dedo y la tendosinovitis. La dactilitis se evaluó en 20 dedos (10 dedos de las manos y 10 dedos de los pies) usando un sistema de puntuación de 0 a 3 (no dactilitis a dactilitis severa). La puntuación de la dactilitis se calculó como la sumatoria de la puntuación de los 20 dedos (rango de 0 a 60). Se considera que un paciente tiene dactilitis en los dedos si la puntuación es > 0. N.S. = No significativa |
|||
La mediana del porcentaje de mejoría de la puntuación de entesitis y dactilitis observadas en la semana 24 se mantuvo hasta la semana 104. En los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de la mediana porcentual de la mejoería de la puntuación de entesitis y dactilitis desde la semana 104 hasta la semana 256.
Estudio por vía IV de APs: El tratamiento con SIMPONI® IV, en comparación con el placebo, produjo una mejoría significativa en los signos y síntomas según lo demostrado por el porcentaje de pacientes con una respuesta ACR 20 en la semana 14 (ver Tabla 26). En el grupo tratado con SIMPONI® IV, el 75% de los pacientes alcanzó una respuesta ACR 20 en comparación con el 22% en el grupo del placebo (p<0.001). Este beneficio fue observado consistentemente cuando se evaluó en los pacientes tratados con SIMPONI® IV en los cinco subtipos de APs. Las respuestas ACR 20 observadas en los grupos tratados con SIMPONI® IV fueron similares en los pacientes que estaban recibiendo y que no estaban recibiendo MTX de manera concomitante. Además, el porcentaje de los pacientes que alcanzaron una respuesta ACR 20 en la semana 24 fue significativamente mayor para los pacientes que recibieron SIMPONI® IV (77%) en comparación con los pacientes que recibieron el placebo (24%) (p<0.001) (Tabla 26).
El porcentaje de los pacientes que alcanzaron ACR 50, ACR 70 y ACR 90 fue mayor en los pacientes tratados con SIMPONI® IV en comparación con el placebo (Tabla 26). El porcentaje de pacientes que alcanzaron una respuesta ACR 50 en la semana 14 fue 44% para los pacientes tratados con SIMPONI® IV y 6.3% para los pacientes tratados con el placebo (p<0.001). En la semana 24, el porcentaje de los pacientes que alcanzaron una respuesta ACR 50 fue 54% y 6.3% (p<0.001) para los grupos con SIMPONI® IV y el placebo, respectivamente. El porcentaje de los pacientes que alcanzaron una respuesta ACR 70 en la semana 14 fue 25% para los pacientes tratados con SIMPONI® IV y 2.1% para los pacientes tratados con el placebo (p<0.001). En la semana 24, el porcentaje de los pacientes que alcanzaron una respuesta ACR 70 fue 33% y 3.3% (p<0.001) para los grupos con SIMPONI® IV y el placebo, respectivamente. El porcentaje de los pacientes que alcanzaron una respuesta ACR 90 en la semana 14 fue 3.7% para los pacientes tratados con SIMPONI® IV y 0.4% para los pacientes tratados con el placebo (p=0.011). En la semana 24, el porcentaje de los pacientes que alcanzaron una respuesta ACR 90 fue 6.2% y 0% (p<0.001) para los grupos con SIMPONI® IV y el placebo, respectivamente (Tabla 26).
|
Tabla 26: Estudio por vía IV de APs: Porcentaje de pacientes con respuestas ACR en la semanas 14 y 24: pacientes aleatorizados |
|||
|
Placebo N=239a |
SIMPONI® IV N=241a |
Diferencia del tratamiento |
|
|
ACR 20 (% de respondedores) |
|||
|
Semana 14 |
22% |
75% |
53%* (45.8. 60.9) |
|
Semana 24 |
24% |
77% |
53%* (44.9, 60.1) |
|
ACR 50 (% de respondedores) |
|||
|
Semana 14 |
6.3% |
44% |
37%* (30.3, 44.2) |
|
Semana 24 |
6.3% |
54% |
47%* (40.2, 54.2) |
|
ACR 70 (% de respondedores) |
|||
|
Semana 14 |
2.1% |
25% |
22%* (16.7, 28.1) |
|
Semana 24 |
3.3% |
33% |
29%* (23.1, 35.8) |
|
ACR 90 (% de respondedores) |
|||
|
Semana 14 |
0.4% |
3.7% |
3.3%** (0.80, 5.8) |
|
Semana 24 |
0 |
6.2% |
6.2* (3.2, 9.3) |
|
a N refleja los pacientes aleatorizados. El texto en negrita indica el criterio de evaluación principal. * p<0.001 ** p=0.011 |
|||
El porcentaje de los pacientes que alcanzaron respuestas ACR 20 por visita hasta la semana 24 para el estudio por vía IV de APs se muestra en la Figura 6. Después de la administración inicial, se observaron respuestas ACR 20 en el 46% de los pacientes tratados con SIMPONI® IV en la primera evaluación (semana 2) en comparación con el 7.5% de los pacientes tratados con el placebo. También se observaron respuestas para ACR 50 y ACR 70 tempranamente en la primera evaluación (semana 2).
Figura 6: Estudio por vía IV de APs: Porcentaje de los pacientes que alcanzaron respuesta ACR 20 hasta la semana 24: Pacientes aleatorizados
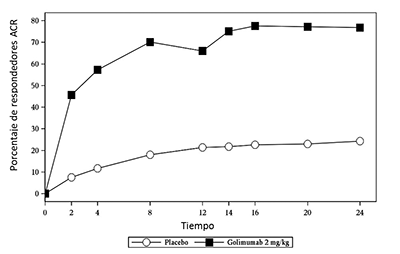
La Tabla 27 muestra la mediana de la mejoría porcentual en los componentes de los criterios de la respuesta ACR para los grupos con SIMPONI® IV y el placebo en el estudio por vía IV de APs.
|
Tabla 27: Estudio por vía IV de APs: Mediana de la mejoría porcentual desde el estado basal en los componentes de ACR en la semana 14 y la semana 24, pacientes aleatorizados |
||
|
Placebo N=239a |
SIMPONI® IV N=241a |
|
|
Componentes de ACR |
||
|
Número de articulaciones inflamadas (0-66) |
||
|
Estado basal |
12 |
11 |
|
Semana 14 |
32% |
87% |
|
Semana 24 |
43% |
90% |
|
Número de articulaciones sensibles (0-68) |
||
|
Estado basal |
24 |
22 |
|
Semana 14 |
15% |
71% |
|
Semana 24 |
23% |
79% |
|
Evaluación del paciente del dolor (0-100 mm) |
||
|
Estado basal |
67 |
67 |
|
Semana 14 |
12% |
53% |
|
Semana 24 |
15% |
62% |
|
Evaluación general del paciente de la actividad de la enfermedad (0-100 mm) |
||
|
Estado basal |
66 |
68 |
|
Semana 14 |
13% |
51% |
|
Semana 24 |
12% |
59% |
|
Evaluación general del médico de la actividad de la enfermedad (0-100 mm) |
||
|
Estado basal |
65 |
64 |
|
Semana 14 |
16% |
70% |
|
Semana 24 |
24% |
75% |
|
Índice de discapacidad (HAQ) (0-3) b |
||
|
Estado basal |
1.2 |
1.2 |
|
Semana 14 |
10% |
44% |
|
Semana 24 |
11% |
50% |
|
PCR (mg/L)c |
||
|
Estado basal |
14 |
12 |
|
Semana 14 |
22% |
86% |
|
Semana 24 |
21% |
83% |
|
Nota: todos los valores son medianas. a N refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar. b Indice de discapacidad del cuestionario de evaluación de la salud. c PCR = Proteina C reactiva |
||
La Tabla 28 muestra la mediana del cambio desde el estado basal en la semana 14 en los componentes del criterio de la respuesta ACR para los grupos con SIMPONI® IV y el placebo en el estudio por vía IV de APs.
|
Tabla 28: Estudio por vía IV de APs: Mediana del cambio desde el estado basal en los componentes de ACR en la semana 14: Sujetos aleatorizados |
||||
|
Placebo N=239a |
SIMPONI® IV N=241a |
|||
|
Estado basal |
Semana 14 cambio desde el estado basal |
Estado basal |
Semana 14 cambio desde el estado basal |
|
|
Componentes de ACR |
||||
|
Número de articulaciones inflamadas (0-66) |
12 |
-4.0 |
11 |
-8.0 |
|
Número de articulaciones sensibles (0-68) |
24 |
-3.0 |
22 |
-13 |
|
Evaluación del paciente del dolor (0-100 mm) |
67 |
-8.0 |
67 |
-33 |
|
Evaluación global del paciente de la actividad de la enfermedad (0-100 mm) |
66 |
-9.0 |
68 |
-33 |
|
Evaluación global del médico de la actividad de la enfermedad (0-100 mm) |
65 |
-11 |
64 |
-39 |
|
Índice de discapacidad (HAQ) (0-3)b |
1.2 |
-0.13 |
1.2 |
-0.63* |
|
PCR (mg/L)c |
14 |
-2.1 |
12 |
-9.2 |
|
Nota: todos los valores son medianas. * p<0.001. a N refleja los pacientes aleatorizados: el número real de pacientes evaluables para cada criterio de evaluación puede variar. b Indice de discapacidad del Cuestionario de evaluación de la salud. c PCR = Proteína C reactiva |
||||
El porcentaje de los pacientes que alcanzaron el criterio de respuesta de artritis psoriásica modificado (mPsARC), la puntiación de la actividad de la enfermedad incluyendo 28 articulaciones utilizando la respuesta de la proteína C reactiva (DAS28) y la remisión DAS28 también fue mayor en los pacientes tratados con SIMPONI® IV en comparación con el placebo en la semana 14 y en la semana 24 (Tabla 29). El porcentaje de los pacientes que alcanzaron una respuesta mPsARC en la semana 14 y en la semana 24 fue 84% para los pacientes tratados con SIMPONI® IV y 34% para los pacientes tratados con el placebo (p<0.001). En el estado basal, la puntuación promedio de DAS28 en el grupo tratado con SIMPONI® IV y en el grupo tratado con el placebo fue 5.4 y 5.5, respectivamente. El porcentaje de pacientes que alcanzaron una respuesta DAS28 en la semana 14 fue 93% para los pacientes tratados con SIMPONI® IV y 40% para los pacientes tratados con el placebo (p<0.001). En la semana 24, el porcentaje de los pacientes que alcanzaron una respuesta DAS28 fue 93% y 40% (p<0.001) para los grupos con SIMPONI® IV y el placebo, respectivamente. El porcentaje de los pacientes que alcanzaron remisión DAS28 en la semana 14 fue 37% para los pacientes tratados con SIMPONI® IV y 4.6% para los pacientes tratados con el placebo (p<0.001). En la semana 24, el porcentaje de los pacientes que alcanzaron remisión DAS28 fue 42% y 7.1% (p<0.001) para los grupos tratados con SIMPONI® IV y el placebo, respectivamente. El cambio promedio desde el estado basal en la puntuación de DAS28 score en la semana 14 fue -2.3 y de -0.6 en los pacientes tratados con SIMPONI® IV y con el placebo, respectivamente. En la semana 24, el cambio promedio desde el estado basal en la puntuación de DAS28 fue -2.50 y -0.7 en los pacientes tratados con SIMPONI® IV y con el placebo, respectivamente. Una puntuación DAS28 negativa o decreciente es indicativa de mejoría. Se observaron respuestas para mPsARC y DAS28 tempranamente enla primera evaluación (semana 2).
|
Tabla 29: Estudio por vía IV de APs: Porcentaje de pacientes con respuesta de PsARC modificada, respuesta de DAS28 y remisión DAS28 en la semanas 14 y 24: Pacientes aleatorizados |
|||
|
Placebo N=239a |
SIMPONI® IV N=241a |
Diferencia de tratamiento (IC del 95%) |
|
|
PsARC modificadob (% con respuesta) |
|||
|
Semana 14 |
34% |
84% |
50% (42.3, 57.6) |
|
Semana 24 |
34% |
84% |
50% (41.9, 57.1) |
|
Respuesta de DAS28 (PCR) (% de respondedores) |
|||
|
Semana 14 |
40% |
93% |
52% (45.3, 59.4) |
|
Semana 24 |
40% |
93% |
53% (46.2, 60.2) |
|
Remisión DAS28 (PCR) (% de remitentes) |
|||
|
Semana 14 |
4.6% |
37% |
32% (25.3, 38.6) |
|
Semana 24 |
7.1% |
42% |
35% (27.8, 41.8) |
|
a N refleja los pacientes aleatorizados b El Criterio de respuesta de artritis psoriátrica modificado (PsARC) se define como una mejoría en al menos dos de los siguientes criterios: • ≥ 30% de disminución en el recuento de articulaciones inflamadas • ≥ 30% de disminución en el recuento de articulaciones sensibles • ≥ 20% de mejoría en la evaluación general del paciente en una escala análoga visual; y • ≥ 20% de mejoría en la evaluación general del médico en una escala analógica visual • Incluyen to por lo menos 1 de los criterios de articulaciones y sin deterioro en los otros criterios. |
|||
La Tabla 30 muestra la mejoría en la entesitis y la dactilitis para los grupos de SIMPONI® IV y el placebo en el estudio por vía IV de APs. Los pacientes con entesitis en el estado basal fueron evaluados para determinar la mejoría utilizando el Índice de Entesitis de Leeds (LEI) en una escala de 0-6. En el estado basal, la mediana de la puntuación de LEI en los pacientes con entesitis en el grupo tratado con SIMPONI® IV y en el grupo tratado con el placebo fue de 3.0 y de 3.0, respectivamente. Los pacientes tratados con SIMPONI® IV mostraron una mediana en la mejoría de la puntuación LEI de 2.0 en comparación con una mediana de la mejoría en los pacientes tratados con el placebo de 0.0 en la Semana 14 (p<0.001). Los pacientes con dactilitis en el estado fueron evaluados para determinar la mejoría utilizando una escala de 0-60. En el estado basal, la mediana de la puntuación de dactilitis en los pacientes con dactilitis en el grupo tratado con SIMPONI® IV y en el grupo tratado con el placebo fue 5.5 y 6.0, respectivamente. Los pacientes tratados con SIMPONI® IV mostraron una mediana de la mejoría en la puntuación de dactilitis de 4.0 en comparación con una mediana de la mejoría de 2.0 en los pacientes tratados con el placebo en la semana 14 (p<0.001). Las mejorías en la entesitis y la dactilitis se mantuvieron en el grupo tratado con SIMPONI® IV hasta la semana 24.
|
Tabla 30: Estudio por vía IV de APs: Mediana de la mejoría dede el estado basal y porcentaje de mejoría en la puntuación de entesitis en pacientes con entesitis en el estado basal y mediana de la mejoría desde el estado basal y porcentaje de la mejoría en la puntuación de dactilitis en pacientes con dactilitis en el estado basal: pacientes aleatorizados |
||
|
Placebo N=239a |
SIMPONI® IV N=241a |
|
|
Puntuación de entesitis (mediana) |
||
|
Pacientes con entesitis en el estado basal |
181 |
185 |
|
Puntuación en el estado basal |
3.0 |
3.0 |
|
Mejoría desde el estado basal |
||
|
Semana 14 |
0.0 |
2.0* |
|
Semana 24 |
1.0 |
2.0 |
|
Porcentaje de mejoría |
||
|
Semana 14 |
0% |
83% |
|
Semana 24 |
33% |
100% |
|
Puntuación de dactilitis (mediana) |
||
|
Pacientes con dactilitis en el estado basal |
124 |
134 |
|
Estado basal |
6.0 |
5.5 |
|
Mejoría desde el estado basal |
||
|
Semana 14 |
2.0 |
4.0* |
|
Semana 24 |
2.0 |
4.0 |
|
Porcentaje de mejoría |
||
|
Semana 14 |
21% |
100% |
|
Semana 24 |
54% |
100% |
|
Nota: Todos los valores son medianas. * p<0.001 a N refleja los pacientes aleatorizados: el número real de pacientes evaluables para cada criterio de evaluación puede variar. Entesitis: El Índice de entesitis de Leeds (LEI) evalúa la entesitis en pacientes con APs, y evalúa la presencia (puntuación de 1) o ausencia (puntuación de 0) del dolor al aplicar presión en las siguientes entesis: epicóndilo lateral de codo (izquierdo y derecho), cóndilo femoral medio (izquierdo y derecho) e inserción del tendón de Aquiles (izquierdo y derecho). Los pacientes fueron calificados sobre una escala de 0-6. Dactilitis: La dactilitis se evaluó en 20 dedos (10 dedos de las manos y 10 dedos de los pies) usando un sistema de puntuación de 0 a 3 (sin dactilitis a dactilitis severa). La puntuación de dactilitis se calculó como la suma de las calificaciones de todos los 20 dedos (rango de 0 a 60). Se considera que un paciente tiene dedos con dactilitis y la puntuación es > 0. |
||
Los pacientes son clasificados considerando si alcanzaron una enfermedad de artritis psoriásica con actividad mínima (MDA) si se cumplen cinco de las siguientes siete medidas de resultado: recuento de articulaciones sensibles ≤1; recuento de articulaciones inflamadas ≤1; PASI ≤1 o ASC ≤ 3; puntuación del paciente con escala analógica visual (VAS) de ≤15; puntuación VAS de la actividad de la enfermedad global del paciente de ≤ 20; puntuación HAQ ≤ 0.5 y puntos de entesis sensibles ≤1. Una mayor proporción de los pacientes en el grupo tratado con SIMPONI® IV en comparación con el placebo alcanzó respuestas de actividad de la enfermedad mínima en la semana 14 (27% frente al 4.2%) y 24 (34% frente al 4.6%).
Respuesta de la psoriasis en la piel y uñas:
Estudio de APs: Entre los pacientes con afectación de la piel por psoriasis ≥ 3% del ASC en el estado basal, un porcentaje significativamente mayor de pacientes alcanzó respuesta PASI 75 en la semanas 14 y la semana 24 cuando fue tratado con SIMPONI® 50 mg en comparación con el placebo. El porcentaje de los pacientes que alcanzaron una respuesta PASI 75 en el grupo de SIMPONI® 50 mg fue mayor que en el grupo del placebo y fue similar en los pacientes que recibieron y que no recibieron MTX concomitantemente. (Tabla 31 y Figura 7).
Figura 7: Estudio de APs: Porcentaje de pacientes con APs que alcanzaron una respuesta PASI 75 hasta la semana 24 en pacientes aleatorizados con afectación de la piel por psoriasis ≥ 3% del ASC en el estado basal
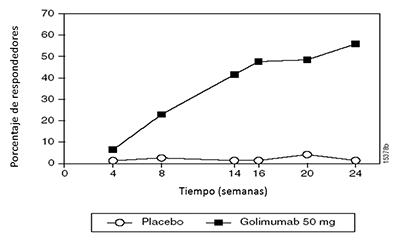
Mayor porcentaje de pacientes tratados con SIMPONI® 50 mg alcanzaron respuestas PASI 50 y PASI 90 en comparación con el placebo y los pacientes tratados con SIMPONI® 50 mg tuvieron mayor porcentaje de mejoría de la puntuación PASI comparados con los pacientes tratados con placebo (Tabla 31). La proporción de los pacientes con respuestas PASI 50, PASI 75 o PASI 90 observada en la semana 24 se mantuvo hasta la semana 104. En los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de respuesta PASI 50, PASI 75 o PASI 90 desde la semana 104 hasa la semana 256.
|
Tabla 31: Estudio de APs: Análisis de eficacia cutánea en pacientes aleatorizados con afectación ≥ 3% del ASC en el estado basal |
||
|
Placebo (N=113)a |
SIMPONI® 50 mg* (N=146)a |
|
|
Pacientes con ≥3% del ASC en el estado basal |
79 |
109 |
|
PASI 50 (% de respondedores) |
||
|
Semana 14 |
10% |
59% |
|
Semana 24 |
8% |
76% |
|
PASI 75 (% de respondedores) |
||
|
Semana 14 |
3% |
40% |
|
Semana 24 |
1% |
56% |
|
PASI 90 (% de respondedores) |
||
|
Semana 14 |
0% |
21% |
|
Semana 24 |
0% |
32% |
|
Puntuación PASI |
||
|
En estado basal |
6.2 |
7.5 |
|
Porcentaje de mejoría |
||
|
Semana 14 |
0% |
64% |
|
Semana 24 |
3% |
79% |
|
*p < 0.001 para PASI 50, PASI 75 y PASI 90; el valor-p no se calculó para el porcentaje de mejoría de la puntuación PASI aN refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar en el punto cronológico. PASI es un índice que se usa para evaluar y calificar la gravedad de las lesiones por psoriasis y su respuesta a la terapia. El índice PASI produce una puntuación numérica que varía de 0 a 72. En el sistema PASI, el cuerpo se divide en 4 regiones: Cabeza (h), tronco (t), extremidades superiores (u) y extremidades inferiores (l), lo cual corresponde a 10%, 30%, 20% y 40% del total del ASC, respectivamente. Cada una de estas áreas se evalúa por separado para eritema, induración y descamación, cada una de las cuales se califica en un rango de 0 a 4 (0 = ninguna, 1 = leve, 2 = moderada, 3 = severa y 4 = muy severa). Cada región también se evalúa para determinar el área afectada de lesiones por psoriasis en un rango de 0 a 6 con 0 como ausencia de área afectada y 6 como área afectada de 90%-100%. |
||
Las evaluaciones de las lesiones objetivo se llevaron a cabo en todos los pacientes independientemente de la extensión de la afectación de la piel por psoriasis y la evaluación general por el médico (PGA) de la uña y el análisis del Índice de severidad de la psoriasis ungüeal (NAPSI) se llevaron a cabo en pacientes con afectación de las uñas en el estado basal. Al margen de la afectación por psoriasis de la ASC en el estado basal, los pacientes tratados con SIMPONI® 50 mg tuvieron una mejoría significativamente mayor de las lesiones psoriásicas objetivo en comparación con el placebo. Además, el porcentaje del cambio desde el estado basal del índice NAPSI y la mejoría de la PGA de las uñas fue significativamente mayor en pacientes tratados con SIMPONI® 50 mg en comparación con el placebo en la semana 14 y en la semana 24 (Tabla 32).
|
Tabla 32: Estudio de APs: Mejoría de las lesiones objetivo y de la PGA de las uñas y del índice NAPSI en pacientes aleatorizados |
||
|
Placebo |
SIMPONI® 50 mg (N =146)a |
|
|
Lesiones objetivo (% de mejoría) |
||
|
Estado basal (mediana) |
6.0 |
6.0 |
|
Semana 14 |
0% |
50%* |
|
Semana 24 |
0% |
6%* |
|
Pacientes con afectación de las uñas en el estado basal |
83 |
95 |
|
PGA de las uñas (%de respondedores) |
||
|
Semana 14 |
14% |
47%* |
|
Semana 24 |
18% |
60%* |
|
NAPSI (% de mejoría) |
||
|
Estado basal (mediana) |
4.0 |
4.0 |
|
Semana 14 |
0% |
25%** |
|
Semana 24 |
0% |
33% * |
|
* p < 0.001 **p = 0.015 aN refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar en el punto cronológico. |
||
La proporción de los pacientes con respuesta PGA de las uñas en la semana 24 se mantuvo hasta la semana 104. La mediana del porcentaje de mejoría de la puntuación del índice NAPSI observada en la semana 24 se mantuvo hasta la semana 104. En los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de respuesta PGA de las uñas y la mediana del porcentaje de mejoría de la puntuación NAPSI desde la semana 104 hasta la semana 256.
Estudio por vía IV de APs: La Tabla 33 muestra la mejoría de la piel psoriásica afectada para los grupos con SIMPONI® IV y el placebo en el estudio por vía IV de APs. Se evaluó la mejoría de los pacientes utilizando el Índice de severidad y área de psoriasis (PASI). En estado basal, la mediana de la puntuación PASI en pacientes con piel psoriásica afectada ≥ 3% del ASC en el grupo tratado con SIMPONI® IV y en el grupo tratado con el placebo fue 6.0 y 8.2, respectivamente. Entre los pacientes con piel afectada con psoriasis ≥ 3% del ASC en el estado basal (82%), un porcentaje significativamente mayor de los pacientes alcanzó una respuesta PASI 75 en la semana 14 cuando fueron tratados con SIMPONI® IV en comparación con el placebo (59% frente a 14%, p<0.001). El porcentaje de los pacientes que alcanzó respuesta PASI 50, PASI 90 y PASI 100 en el grupo tratado con SIMPONI® IV fue mayor que en el grupo tratado con el placebo (Tabla 33).
|
Tabla 33: Estudio por vía IV de APs: Porcentaje de pacientes que alcanzaron respuesta PASI, mediana de la mejoría desde el vaor basal y mejoría porcentual desde el estado basal en la puntuación PASI en la semanas 14 y 24; sujetos aleatorizados con piel afectada con psoriasis ≥ 3% del ASC en el estado basal |
||
|
Placebo N=239a |
SIMPONI® IV N=241a |
|
|
Pacientes con ≥ 3% del ASC en el estado basal |
198 |
196 |
|
PASI 50b (% de respondedores) |
||
|
Semana 14 |
22% |
78% |
|
Semana 24 |
29% |
80% |
|
PASI 75b (% de respondedores) |
||
|
Semana 14 |
14% |
59%* |
|
Semana 24 |
13% |
65% |
|
PASI 90b (% de respondedores) |
||
|
Semana 14 |
6.6% |
39% |
|
Semana 24 |
7.6% |
43% |
|
PASI 100 b (% de respondedores) |
||
|
Semana 14 |
4.5% |
17% |
|
Semana 24 |
5.6% |
26% |
|
Puntuación PASI b (Mediana) |
||
|
Estado basal |
6.0 |
8.2 |
|
Mejoría desde el estado basal (Mediana) |
||
|
Semana 14 |
0.3 |
5.5 |
|
Semana 24 |
0.9 |
5.3 |
|
Mejoría porcentual (Mediana) |
||
|
Semana 14 |
8.5% |
85% |
|
Semana 24 |
22% |
88% |
|
a N refleja los pacientes asignados aleatorizados: el número real de pacientes evaluables para cada criterio de evaluación puede variar en cada punto de valoración. * p<0.001 b El Índice de severidad y área de psoriasis (PASI) es un índice que se utiliza para evaluar y calificar la severidad de las lesiones psoriásicas y su respuesta a la terapia. El PASI produce una puntuación numérica que varía de 0 a 72. En el sistema PASI, el cuerpo se divide en cuatro regiones: la cabeza (h), el tronco (t), las extremidades superiores (u) y las extremidades inferiores (l), que representan el 10%, 30%, 20% y 40% del ASC total, respectivamente. Cada una de estas áreas es evaluada por separado para determinar eritema, induración y descamación, cada uno de los cuales es clasificado utilizando una escala de 0 a 4 (0 = ninguno, 1 = leve, 2 = moderado, 3 = severo, y 4 = muy severo). Cada región también es evaluada por el área afectada por lesiones psoriásicas utilizando una escala de 0 a 6 donde 0 es no afectado y 6 afectado de 90% - 100%. Una respuesta PASI 50 se define como una mejoría ≥ 50% en la puntuación PASI desde el estado basal; la respuesta PASI 75, PASI 90 y PASI 100 se definen de manera similar. |
||
La Tabla 34 muestra la mejoría de las uñas afectadas para los grupos con SIMPONI® IV y el placebo en el estudio por vía IV de APs. La mejoría de los pacientes se evaluó utilizando el Índice de la severidad de la psoriasis üngueal modificado (mNAPSI). El mNAPSI se utiliza para evaluar la afectación de las uñas psoriásicas en pacientes con artritis psoriásica. Cada uña se evalúa sobre un grupo de características, con una puntuación de 0-13 y una puntuación total de 0-130 para cada paciente. En el estado basal, la mediana de la puntuación mNAPSI en los pacientes con uñas afectadas en el grupo tratado con SIMPONI® IV y en el grupo tratado con el placebo fue 13 y 15, respectivamente. Entre los pacientes con uñas afectadas en el estado basal (76%), la mediana de la mejoría desde el estado basal en la puntuación mNAPSI fue mayor en los pacientes tratados con SIMPONI® IV en comparación con los pacientes tratados con el placebo en la semana 14 (6.0 frente a 0.0, respectivamente). La mejoría porcentual desde el estado basal en la puntuación mNAPSI fue mayor en el grupo con SIMPONI® IV que en el grupo del placebo.
|
Tabla 34: Estudio por vía IV de APs: Mediana de la mejoría desde el estado basal y mejoría porcentual en la puntuación mNAPSI en pacientes con las uñas afectadas en el estado basal; pacientes aleatorizados |
||
|
Placebo N=239a |
SIMPONI® IV N=241a |
|
|
Pacientes con uñas afectadas en el estado basal |
170 |
197 |
|
Puntuación mNAPSI (Mediana)b |
||
|
Estado basal |
15 |
13 |
|
Mejoría desde el estado basal (Mediana) |
||
|
Semana 14 |
0.0 |
6.0 |
|
Semana 24 |
2.0 |
7.0 |
|
Mejoría porcentual (Mediana) |
||
|
Semana 14 |
0.0 |
63% |
|
Semana 24 |
14% |
76% |
|
a N refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar en el tiempo de evaluación. b El Índice de severidad de psoriasis ungüeal modificado (mNAPSI) se utiliza para evaluar las uñas psoriásicas afectadas. Se evaluaron tres características o grupos de características (picaduras, onicolisis y discromia por gota de aceite y disgregación) de cada uña utilizando una escala de 0 a 3. Se evaluaron cuatro características (leuconiquia, hemorragias por astilla, hiperqueratosis y puntos rojos en la lúnula) ya sea como presentes o ausentes para cada uña. Cada uña se evaluó sobre un grupo de características, con una puntuación de 0-13, y una puntuación total de 0-130 para cada paciente. |
||
El cuestionario del Índice de calidad de vida dermatológico (DLQI) es un instrumento de la calidad de vida específico en dermatología utilizado para evaluar la perspectiva del paciente sobre el impacto de los trastornos de la piel sobre la vida diaria. La puntuación total del índice DLQI varía de 0-30, donde las puntuaciones bajas indican menor impacto de la psoriasis en la vida del paciente. En una enfermedad dermatológica, se ha determinado que un cambio de 5 puntos tiene relevancia. En el estado basal, el 59% de los pacientes presentaron una puntuación DLQI > 1 y una piel psoriásica afectada ≥ 3% del ASC. En el estado basal, la mediana de la puntuación DLQI en el grupo tratado con SIMPONI® IV y en el grupo tratado con el placebo entre esos pacientes fue 13 y 12, respectivamente. Entre estos pacientes, la mediana de la disminución (mejoría) desde el estado basal en la puntuación DLQI fue mayor en los pacientes tratados con SIMPONI® IV que en el grupo del placebo en la semana 14 (-6.0 frente a -1.0, respectivamente) y en la semana 24 (-7.0 frente a -1.0, respectivamente).
Respuesta radiográfica:
Estudio de APs: El daño estructural en ambas manos y pies se evaluó radiograficamente por medio del cambio desde el estado basal de la puntuación van der Heijde – Sharp (vdH-S), modificada para APs mediante la adición de las articulaciones interfalángicas distales (DIP) de la mano. En la semana 24, SIMPONI® 50 mg inhibió significativamente la progresión del daño estructural en comparación con el placebo. Los resultados se muestran en la Tabla 35. Los pacientes tratados con SIMPONI® con o sin MTX tuvieron menos progresión que los pacientes que recibieron placebo con o sin MTX.
|
Tabla 35: Estudio en APs: Cambio radiológico desde el estado basal (semana 24) |
||
|
Placebo (N=113)a |
SIMPONI® 50 mg (N=146)a |
|
|
Puntuación total |
||
|
Estado basal |
18.2 (27.8) |
23.9 (35.4) |
|
Cambio desde el estado basal |
0.27 (1.3) |
-0.16 (1.3)* |
|
Puntuación de la erosión |
||
|
Estado basal |
10.6 (16.1) |
13.7 (19.6) |
|
Cambio desde el estado basal |
0.32 (0.9) |
- 0.09 (0.9)** |
|
Puntuación JSN |
||
|
Estado basal |
7.5 (12.5) |
10.1 (16.8) |
|
Cambio desde el estado basal |
-0.03 (0.7) |
-0.03 (0.6) |
|
a N refleja los pacientes aleatorizados, el número real de los pacientes para cada análisis puede variar * p =0.011. **p<0.001 Los valores son medias (desviación estándar) de la puntuación total vdH-S modificada para APs. |
||
Un número significativamente mayor de pacientes del grupo de SIMPONI® 50 mg no tuvo nuevas erosiones o nuevo estrechamiento de espacio articular (JSN) en las articulaciones que no estaban afectadas en el estado basal en comparación con el placebo (Tabla 36).
|
Tabla 36: Estudio de APs: Nuevas erosiones y JSN en articulaciones previamente no afectadas (semana 24) |
|||
|
Placebo (N=113)a |
SIMPONI® 50 mg (N=146)a |
Valor-p |
|
|
Pacientes por lo menos con 1 articulación previamente no afectada |
102 |
132 |
|
|
Pacientes sin nuevas erosiones |
73 (72%) |
115 (87%) |
0.003 |
|
Pacientes por lo menos con 1 articulación previamente no afectada |
102 |
132 |
|
|
Sujetos sin nuevo JSN |
90 (88%) |
128 (97%) |
0.008 |
|
a N refleja los pacientes aleatorizados. Los valores son números (%) |
|||
Hubo un número significativamente mayor de sujetos en el grupo de SIMPONI® 50 mg que no presentaron incremento desde el estado basal de la puntuación total vdH-S modificada para APs en comparación con el grupo del placebo (Tabla 37).
|
Tabla 37: Estudio de APs: Número de pacientes con cambio desde el estado basal de la puntuación total vdH-S modificada para APs ≤ 0 (Semana 24) |
|||
|
Placebo (N=102) |
SIMPONI® 50 mg (N=132) |
Valor-p |
|
|
Pacientes con cambio desde el estado basal de la puntuación total vdH-S modificada para APs ≤ 0 |
64 (63%) |
104 (79%) |
0.007 |
|
Los valores son números (%) |
|||
El efecto de SIMPONI® sobre la progresión radiográfica se mantuvo en la semana 104. De los 114 pacientes aleatorizados para recibir SIMPONI® 50 mg que continuaron el tratamiento con SIMPONI® después de la semana 52, el 77% tuvo un cambio desde el estado basal de la puntuación total vdH-S modificada para APs ≤ 0 en la semana 104. Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observó un efecto similar en la progresión radiográfica dede la semana 104 hasta la semana 256.
Estudio por vía IV de APs: El daño estructural de las articulaciones fue evaluado radiográficamente y expresado como un cambio en la puntuación total van der Heijde-Sharp (vdH-S) modificada y sus componentes, la puntuación de la erosión y la puntuación del estrechamiento del espacio articular (JSN) en la semana 24 en comparación con el estado basal. En el estado basal, la puntuación vdH-S total promedio modificada fue 35.0, la puntuación de la erosión promedio fue 21.7 y la puntuación promedio del JSN fue 13.3. Ningún cambio desde el estado basal o una disminución desde el estado basal en las puntuaciones vdH-S total modificado, de la erosión o del JSN es indicativo de inhibición de la progresión. En la semana 24, SIMPONI® IV inhibió significativamente la progresión del daño estructural en comparación con el placebo, según la evaluación mediante la puntuación vdH-S total modificada y sus componentes, como se muestra en la Tabla 38.
|
Tabla 38: Estudio 1 por vía IV de APs 1: Cambio radiográfico desde el estado basal en la semana 24 |
||||
|
Placebo N=237 |
SIMPONI® IV N=237 |
|||
|
Estado basal |
Cambio en la semana 24 desde el estado basal |
Estado basal |
Cambio en la semana 24 desde el estado basal |
|
|
Puntuación vdH-S total modificada |
34.4 (53.5) |
2.0 (0.3) |
35.5 (55.1) |
-0.4 (0.1)* |
|
Puntuación de la erosión |
21.1 (30.2) |
1.3 (2.8) |
22.2 (31.7) |
-0.3 (1.7)* |
|
Puntuación del JSN |
13.3 (24.3) |
0.6 (1.7) |
13.2 (24.8) |
-0.1 (1.1)* |
|
Nota: todos los valores son promedios (desviación estándar). * p<0.001 |
||||
Entre los pacientes con ≥ 1 articulación con una puntuacipón del JSN de 0 en el estado basal (92%), una mayor proporción de los pacientes tratados con SIMPONI® IV no mostraron erosiones nuevas ni estrechamientos de los espacios articulares nuevos en comparación con los pacientes tratados con el placebo en la semana 24 (ver Tabla 39).
|
Tabla 39: Estudio por vía IV de APs: Porcentaje de los pacientes con erosiones nuevas y JSN en articulaciones no afectadas previamente (semana 24) |
|||
|
Placebo N=237 |
SIMPONI® IV N=237 |
Valor de p |
|
|
Pacientes con por lo menos 1 articulación no afectada previamente |
213 |
230 |
|
|
Pacientes sin erosiones nuevas |
115 (54%) |
171 (74%) |
<0.001 |
|
Pacientes con por lo menos 1 articulación no afectada previamente |
214 |
229 |
|
|
Pacientes sin JSN nuevos |
162 (76%) |
205 (90%) |
<0.001 |
|
Los valores son números (%). |
|||
En la semana 24, una mayor proporción de los pacientes en el grupo tratado con SIMPONI® IV (72%) no presentó progresión del daño estructural (cambio en la puntuación vdH-S total modificada ≤ 0), en comparación con el 43% de los pacientes en el grupo del placebo (p<0.001) (Tabla 40).
|
Tabla 40: Estudio por vía IV de APs: Número de pacientes con un cambio desde el estado basal en la puntuación vdH-S total modificada para APs ≤ 0 (semana 24) |
|||
|
Placebo N=237 |
SIMPONI® IV N=237 |
Valor de p |
|
|
Pacientes con cambio desde el estado basal en la puntuación vdH-S total modificada para APs ≤ 0 |
102 (43%) |
170 (72%) |
<0.001 |
|
Los valores son números (%). |
|||
Mejoría del funcionamiento físico y la calidad de vida relacionada con la salud:
Estudio de APs: El funcionamiento físico y la calidad de vida relacionada con la salud se evaluaron usando el índice de discapacidad del HAQ y el cuestionario de salud SF-36. En el estudio en APs, los pacientes tratados con SIMPONI® 50 mg mostraron una mediana significativamente mayor de la mejoría del HAQ en comparación con el placebo en la semana 14 (0.25 frente a 0.00; p<0.001) y en la semana 24 (0.25 frente a 0.00; p<0,001) (Tabla 41). La media (± SD) de la mejoría del HAQ desde el estado basal hasta la semana 14 fue 0.31 ± 0.504 en el grupo tratado con SIMPONI® y 0.04 ± 0.441 en el grupo del placebo. La media de la mejoría desde el estado basal hasta la semana 24 fue 0.33 ± 0.552 en el grupo de SIMPONI® y –0.01 ± 0.488 en el grupo del placebo.
El porcentaje de los pacientes que alcanzaron mejoría clínicamente significativas del HAQ de ≥ 0.25 y ≥ 0.30 en el cambio desde el estado basal también fue significativamente mayor en los pacientes que recibieron SIMPONI® 50 mg en comparación con el placebo.
|
Tabla 41: Estudio de APs: Mejoría del funcionamiento físico medida por el HAQ en pacientes aleatorizados |
||
|
Placebo |
SIMPONI® 50 mg* (N=146)a |
|
|
Puntuación HAQ en el estado basal |
||
|
Media ± SD |
1.03 ± 0.548 |
0.98 ± 0.648 |
|
Mediana |
1.00 |
1.00 |
|
Mejoría del HAQ |
||
|
Semana 14 |
||
|
Media ± SD |
0.04 ± 0.441 |
0.31 ± 0.504 |
|
Mediana |
0.00 |
0.25 |
|
Semana 24 |
||
|
Media ± SD |
-0.01 ± 0.488 |
0.33 ± 0.552 |
|
Mediana |
0.00 |
0.25 |
|
HAQ ≥ 0.25 de mejoría (% de respondedores) |
||
|
Media ± DE |
35% |
53% |
|
Mediana |
29% |
50% |
|
HAQ ≥ 0.3 de mejoría (% de respondedores) |
||
|
Media ± SD |
26% |
39% |
|
Mediana |
22% |
43% |
|
*p < 0.05 para todas las comparaciones; los cálculos del valor-p se basan en comparaciones de los valores de la mediana para criterios continuos. aN refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar en el punto cronológico. |
||
Cincuenta y tres por ciento de los pacientes aleatorizados al grupo de SIMPONI® 50 mg alcanzaron una mejoría ≥ 0.3 del HAQ desde el estado basal hasta la semana 104. En los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de mejoría del HAQ DI desde la semana 104 hasta la semana 256.
En el estudio en APs, los pacientes tratados con SIMPONI® 50 mg mostraron una mediana significativamente mayor de la mejoría desde el estado basal de la puntuación del resumen del componente físico (PCS) del SF-36 en comparación con el placebo en la semana 14 (5.5 frente a 0.0; p<0.001) y en la semana 24 (6.6 frente a 0.0; p<0.001). La media del cambio desde el estado basal de la puntuación del PCS del SF-36 en la semana 14 fue mayor en el grupo de SIMPONI® 50 mg que en el grupo del placebo (6.5 ± 8.9 frente a 0.6 ± 7.7, respectivamente). La media de la mejoría del funcionamiento físico del PCS del SF-36 se mantuvo hasta la semana 24 (Tabla 42). La mejoría del PCS del SF-36 observada en la semana 24 se mantuvo hasta la semana 104. En los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de mejoría del PCS del SF-36 desde la semana 104 hasta a la semana 256.
Los pacientes tratados con SIMPONI® 50 mg mostraron una mediana significativamente mayor de la mejoría desde el estado basal de la puntuación del resumen del componente mental (MCS) del SF-36 en comparación con el placebo en la semana 14 (2.0 frente a -0.6; p=0.044) y en la semana 24 (1.9 frente a –1.0; p=0.005). La media del cambio desde el estado basal de la puntuación del MCS del SF-36 en la semana 14 fue mayor en el grupo de SIMPONI® 50 mg que en el grupo del placebo (2.8 ± 10.3 frente a 0.4 ± 11.4, respectivamente). La media de la mejoría de la función mental del MCS de SF-36 se mantuvo hasta la semana 24 (Tabla 42). La mejoría del MCS del SF-36 observada en la semana 24 se mantuvo hasta la semana 104. En los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de mejoría del MCS del SF-36 desde la semana 104 hasta la semana 256.
|
Tabla 42: Estudio de APs: Mejoría de la calidad de vida relacionada con la salud medida por SF-36 en pacientes aleatorizados |
|||
|
Placebo |
SIMPONI® 50 mg (N=146)a |
Valor-p* |
|
|
Puntuación del PCS del SF-36 en el estado basal |
|||
|
Media ± SD |
31.9 ± 9.3 |
33.0 ± 10.7 |
|
|
Mediana |
30.4 |
31.3 |
|
|
Mejoría del PCS del SF-36 |
|||
|
Semana 14 |
|||
|
Media ± SD |
0.6 ± 7.7 |
6.5 ± 8.9 |
|
|
Mediana |
0.0 |
5.5 |
<0.001 |
|
Semana 24 |
|||
|
Media ± SD |
0.7 ± 8.7 |
7.4 ± 9.2 |
|
|
Mediana |
0.0 |
6.6 |
<0.001 |
|
Puntuación del PCS del SF-36 en el estado basal |
|||
|
Media ± DE |
47.6 ± 10.7 |
45.4 ± 12.2 |
|
|
Mediana |
48.5 |
45.8 |
|
|
Mejoría del PCS del SF-36 |
|||
|
Semana 14 |
|||
|
Media ± SD |
0.4 ± 11.4 |
2.8 ± 10.3 |
|
|
Mediana |
-0.6 |
2.0 |
0.044 |
|
Semana 24 |
|||
|
Media ± SD |
0.6 ± 12.1 |
3.4 ± 10.5 |
|
|
Mediana |
-1.0 |
1.9 |
0.005 |
|
* Los cálculos del valor-p se basan en comparaciones de los valores de la mediana a N refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar en el punto cronológico. |
|||
Estudio por IV de APs: Se evaluaron la función física y la calidad de vida relacionada con la salud utilizando el Índice de discapacidad del HAQ (HAQ-DI), el Cuestionario de salud SF-36, la puntuación del Indice EQ 5D-5L (Cuestionario de Dimensiones EuroQoL 5), la puntuación de la VAS EQ y el Cuestionario sobre fatiga para la evaluación funcional en el tratamiento de enfermedades crónicas (FACIT-Fatiga).
Los pacientes tratados con SIMPONI® IV mostraron una mediana significativamente mayor de la mejoría desde el estado basal en la puntuación HAQ-DI en comparación con el placebo en la semana 14 (0.63 frente a 0.13; p<0.001) y en la semana 24 (0.63 frente a 0.13; p<0.001). La mejoría en la función física según lo evaluado mediante HAQ-DI demostró que la proporción de los pacientes que alcanzaron mejoría clínicamente relevante de ≥ 0.3 en la puntuación HAQ-DI desde el estado basal fue mayor en el grupo tratado con SIMPONI® IV en comparación con el placebo en la semana 14 (69% frente al 32%; p<0.001) y en la semana 24 (69% frente al 33%; p<0.001) (ver Tabla 43).
|
Tabla 43: Estudio por vía IV de APs: Mejoría en la función física medido por el HAQ; pacientes aleatorizados |
||
|
Placebo N=236a |
SIMPONI® IV N=237a |
|
|
Puntuación HAQ en el estado basal |
||
|
Promedio ± DE |
1.26 ± 0.58 |
1.31 ± 0.56 |
|
Mediana |
1.25 |
1.25 |
|
Mejoría desde el estado basal |
||
|
Semana 14 |
||
|
Promedio ± DE |
0.12 ± 0.47 |
0.60 ± 0.53 |
|
Mediana |
0.13 |
0.63 |
|
Semana 24 |
||
|
Promedio ± DE |
0.14 ± 0.49 |
0.63 ± 0.54 |
|
Mediana |
0.13 |
0.63 |
|
Mejoría en HAQ ≥ 0.3 (% respondedores) |
||
|
Semana 14 |
32% |
69% |
|
Semana 24 |
33% |
69% |
|
Mejoría en HAQ ≥ 0.35 (% respondedores) |
||
|
Semana 14 |
32% |
69% |
|
Semana 24 |
33% |
69% |
|
a N refleja los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar por punto de evaluación. b El Índice de discapacidad del Cuestionario de Evaluación de la salud evaluó el grado de dificultad en 8 áreas funcionales (vestirse, pararse, comer, caminar, higiene, alcanzar, agarrar y actividades de la vida diaria) y es calificado desde 0 (ninguna dificultad) hasta 3 (incapacidad para realizar una labor) en donde puntuaciones más bajas indican un mejor funcionamiento. En APs, se ha determinado que una mejoría en la puntuación de 0.30 indica una mejoría clínicamente relevante. |
||
Los pacientes tratados con SIMPONI® IV mostraron una mediana significativamente mayor de la mejoría desde el estado basal en la puntuación del resumen del componente físicos (PCS) del SF-36 en comparación con el placebo en la semana 14 (8.7 frente al 1.8; p<0.001) y en la semana 24 (9.3 frente al 1.5). En el estado basal, la mediana de las puntuaciones de PCS del SF-36 fue 33 en el grupo con SIMPONI® IV y de 34 para el grupo del placebo. Proporciones mayores de sujetos en el grupo con SIMPONI® IV en comparación con el placebo alcanzaron un cambio clínicamente significativo desde el estado basal en la calificación del PCS del SF-36 (un incremento de 5 o más unidades) desde la semana 8 (63% frente al 26%) hasta la semana 24 (70% frente al 29%).
Los pacientes tratados con SIMPONI® IV mostraron una mediana significativamente mayor de la mejoría desde el estado basal en la puntuación del resumen del componente mental (MCS) del SF-36 en comparación con el placebo en la semana 14 (5.2 frente al 0.4; p<0.001) y en la semana 24 (4.1 frente al 1.3). En el estado basal, la mediana de las puntuaciones del MCS del SF-36 fue 43 en el grupo con SIMPONI® IV y de 42 para el grupo del placebo. Proporciones mayores de sujetos en el grupo con SIMPONI® IV en comparación con el placebo alcanzaron un cambio clínicamente significativo desde el estado basal en la puntuación del MCS del SF-36 (un incremento de 5 o más unidades) desde la semana 8 (46% frente al 27%) hasta la semana 24 (47% frente al 29%).
Además, los ocho aspectos del SF-36 que comprenden las puntuaciones PCS y MCS mostraron medianas más grandes de la mejoría en la semana 8, semana 14 y semana 24 para el grupo con SIMPONI® IV en comparación con el grupo del placebo.
EuroQoL 5D-5L es una medida del estado de salud que proporciona una medida de la salud para evaluaciones clínicas y económicas. EQ-5D-5L consta de 2 elementos: el sistema descriptivo EQ-5D-5L y la escala analógica visual EQ (EQ VAS). El sistema descriptivo EQ-5D incluye las siguientes 5 áreas: movilidad, autocuidado, actividades usuales, dolor/malestar y ansiedad/depresión. El EQ VAS registra la salud autoevaluada por el respondedor sobre una VAS, con los criterios de evaluación de “la peor salud imaginable” y “la mejor salud imaginable”.
Los pacientes tratados con SIMPONI® IV alcanzaron mejoría desde el estado basal en la mediana de la puntuación del Índice EQ-5D-5L y la puntuación EQ-VAS en comparación con los pacientes tratados con el placebo en la semana 14 (0.14 frente al 0.02 y 20 mm frente al 4.0 mm, respectivamente). La mediana de los valores en el estado basal para la puntuación del Índice EQ-5D-5L y la puntuación EQ VAS, que fueron comparables entre los grupos del placebo y SIMPONI® IV para todos los sujetos, fueron de 0.6 y 48 mm, respectivamente (ver Tabla 44).
|
Tabla 44: Estudio por vía IV de APs: Calidad de vida relacionada con la salud: pacientes aleatorizados |
||
|
Placebo (N=239)a |
SIMPONI® IV (N=241)a |
|
|
PCS del SF-36 |
||
|
Puntuación en el estado basal |
||
|
Media ± DE |
34 ± 7.2 |
33 ± 6.9 |
|
Mediana |
34 |
33 |
|
Semana 14, mejoría desde el estado basal |
||
|
Media ± DE |
2.7 ± 5.9 |
8.7 ± 7.6 |
|
Mediana |
1.8 |
8.7 |
|
Semana 24, mejoría desde el estado basal |
||
|
Media ± DE |
2.4 ± 6.1 |
9.4 ± 8.1 |
|
Mediana |
1.5 |
9.3 |
|
MCS del SF-36 |
||
|
Puntuación en el estado basal |
||
|
Media ± DE |
4.2 ± 10.2 |
4.3 ± 11.4 |
|
Mediana |
42 |
43 |
|
Semana 14, mejoría desde el estado basal |
||
|
Media ± DE |
1.0 ± 7.6 |
5.3 ± 10.0 |
|
Mediana |
0.43 |
5.2 |
|
Semana 24, mejoría desde el estado basal |
||
|
Media ± DE |
0.8 ± 7.4 |
5.3 ± 10.2 |
|
Mediana |
1.3 |
4.1 |
|
Puntuación del Índice EQ-5Db |
||
|
Puntuación en el estado basalb |
||
|
Media ± DE |
0.59 ± 0.15 |
0.58 ± 0.14 |
|
Mediana |
0.62 |
0.60 |
|
Semana 14, mejoría desde el estado basal |
||
|
Media ± DE |
0.03 ± 0.12 |
0.15 ± 0.16 |
|
Mediana |
0.02 |
0.14 |
|
Semana 24, mejoría desde el estado basal |
||
|
Media ± DE |
0.04 ± 0.13 |
0.16 ± 0.15 |
|
Mediana |
0.03 |
0.14 |
|
EQ-5D VAS (mm) |
||
|
Puntuación en el estado basal (mm)b |
||
|
Media ± DE |
46.2 ± 20.3 |
46.9 ± 20.1 |
|
Mediana |
47.5 |
48 |
|
Semana 14, mejoría desde el estado basal (mm) |
||
|
Media ± DE |
5.3 ± 21.0 |
19 ± 24.3 |
|
Mediana |
4.0 |
20 |
|
Semana 24, mejoría desde el estado basal (mm) |
||
|
Media ± DE |
5.5 ± 23.1 |
20 ± 24.2 |
|
Mediana |
4.0 |
20 |
|
a N refleja a los pacientes aleatorizados: el número real de pacientes evaluables para cada criterio de evaluación puede variar por tiempo de evaluación. b EuroQoL 5D-5L es una medida del estado de salud que prorporciona una medida de la salud para evaluaciones clínicas y económicas. EQ-5D-5L consta de 2 elementos: El sistema descriptivo EQ-5D-5L y la escala analógica visual EQ (EQ VAS). El sistema descriptivo EQ-5D comprende los siguientes 5 aspectos: movilidad, autocuidado, actividades usuales, dolor/malestar y ansiedad/depresión. EQ VAS registra la salud autoevaluada por el respondedor en una escala analógica visual (VAS, mm), con los criterios de evaluación “la peor salud imaginable” y “ la mejor salud imaginable”. |
||
El Cuestionario sobre fatiga para la evaluación funcional en el tratamiento de enfermedades crónicas (FACIT-F) incluye 13 preguntas para evaluar el nivel de fatiga y cansancio de un sujeto en los últimos 7 días en una escala de 0-52. En el estado basal, la mediana de la puntuación FACIT-Fatiga en el grupo tratado con SIMPONI® IV y en el grupo tratado con el placebo fue 28. La mediana de la mejoría desde el estado basal en la puntuación FACIT-Fatiga fue significativamente mayor para SIMPONI® IV en comparación con el grupo del placebo en la semana 14 (8.0 frente al 2.0; p<0.001) y en la Semana 24 (8.0 frente al 3.0; p<0.001) (ver Tabla 45). La mediana del cambio desde el estado basal en FACIT-Fatiga hasta la semana 14 y la semana 24 en el grupo de SIMPONI® IV fue ≥ 4 puntos, lo cual inidica una mejoría clínicamente relevante de la fatiga. Además, una mayor proporción de los pacientes tratados con SIMPONI® IV alcanzaron una mejoría clínicamente relevante (≥ 4 puntos) desde el estado basal para la puntuación FACIT-Fatiga en comparación con el placebo en la semana 14 y 24 (68% frente al 41% y 70% frente al 43%, respectivamente).
|
Tabla 45: Estudio por vía IV de APs: Mejoría en la fatiga medida por FACIT-F; pacientes aleatorizados |
||
|
Placebo N=239a |
SIMPONI® IV N=241a |
|
|
Puntuación FACIT-F en el estado basalb |
||
|
Media ± DE |
27.7 ± 9.7 |
27.9 ± 9.6 |
|
Mediana |
28.0 |
28.0 |
|
Semana 14, mejoría desde el estado basal |
||
|
Media ± DE |
2.2 ± 7.6 |
8.4 ± 9.9 |
|
Mediana |
2.0 |
8.0 |
|
Semana 24, mejoría desde el estado basal |
||
|
Media ± DE |
2.3 ± 7.8 |
9.2 ± 9.8 |
|
Mediana |
3.0 |
8.0 |
|
a N refleja a los pacienets aleatorizados: el número real de pacientes evaluables para este criterio de valoración puede variar por tiempo de evaluación. b El cuestionario sobre fatiga para la evaluación funcional de la terapia de la enfermedad crónica (FACIT-F) consta de 13 preguntas que evalúan el nivel de fatiga y cansancio de un sujeto en los últimos 7 días medido por el cambio promedio desde el estado basal en una escala de 0-52 |
||
Economía sanitaria:
Estudio de APs: Los datos de economía sanitaria sobre utilización de recursos sanitarios, tiempo laboral perdido por los pacientes y cuidadores, empleabilidad y productividad del paciente en el trabajo, el colegio o el hogar se recogieron por medio de cuestionarios aplicados en el estado basal y posteriormente cada 8 semanas. Para evaluar la productividad, se les solicitó a los pacientes que indicaran cuánto afectó su enfermedad su productividad en el trabajo, el colegio o el hogar en las cuatro semanas previas usando una escala analógica visual (VAS) de 0 a 10 cm (de no afectada a sumamente afectada). A continuación se presentan solamente aquellas evaluaciones en las cuales se observaron diferencias estadísticamente significativas entre SIMPONI® 50 mg y el placebo.
En el estado basal, el grupo de SIMPONI® 50 mg y el grupo del placebo tuvieron puntuación comparable de la mediana de la productividad (5.4 frente a 5.5, respectivamente). La mediana de la mejoría en el autoreporte sobre la productividad fue significativamente mayor en el grupo de SIMPONI® 50 mg que en el grupo del placebo en la semana 16 (-1.1 frente a 0.0; p < 0.001) y en la semana 24 (-1.2 frente a 0.0; p < 0.001). La media de la mejoría en el autoreporte sobre la productividad en el grupo de SIMPONI® 50 mg y el grupo del placebo en la semana 16 y en la semana 24 fue de -1.69 ± 2.77 frente a -0.01 ± 2.56 y -1.86 ± 2.74 frente a -0.08 ± 2.62, respectivamente.
También hubo diferencias significativas en el tiempo laboral perdido por los cuidadores entre el grupo de SIMPONI® 50 mg y el grupo del placebo en la semana 16 (p=0.034) y en la semana 24 (p=0.047). La media del tiempo laboral perdido por los cuidadores en el grupo de SIMPONI® 50 mg y en el grupo del placebo en la semana 16 y en la semana 24 fue de 0.16 días ± 0.84 frente a 0.77 ± 2.69 y 0.19 días ± 0.96 frente a 1.11 ± 3.98, respectivamente.
Estudio por vía IV de APs: Se recogieron datos de economía sanitaria sobre productividad del paciente en el trabajo a través del Cuestionario de Limitaciones Laborales (WLQ) y la escala analógica visual (VAS) de productividad en el estado basal y en la semana 8, 14 y 24.
El WLQ es un cuestionario para evaluar la pérdida de productividad laboral relacionada con la salud. Se solicitó a los pacientes que trabajaban tiempo completo o medio tiempo o de manera voluntaria que calificaran su nivel de dificultad o la capacidad para realizar tareas laborales específicas a fin de evaluar la pérdida de productividad laboral relacionada con la salud a través del cuestionario en una escala de 0 (sin limitaciones en ningún momento) a 100 (limitado todo el tiempo). Posteriormente, las puntuaciones de la escala se convirtieron a una puntuación de pérdida de productividad que indica un porcentaje estimado de pérdida de productividad laboral por motivos de salud. En el estado basal, la mediana de la puntuación de la pérdida de productividad WLQ en el grupo tratado con SIMPONI® IV y en el grupo tratado con el placebo fue 8.8 y 8.9, respectivamente. La disminución de la mediana (mejoría) desde el estado basal en la puntuación de la pérdida de productividad WLQ en los grupos con SIMPONI® IV y el placebo en la semana 14 y 24 fue -2.9 frente al -0.45 (p<0.001), y en la semana 24 fue -3.2 frente al -0.7 (p<0.001).
Se solicitó a los pacientes que indicaran el grado en que su enfermedad impactó su productividad diaria usando una escala VAS de 0 a 10 (sin impacto en la productividad (0) a un impacto alto en la productividad (10)). En el estado basal, la mediana del impacto de la enfermedad en la productividad diaria (VAS) en el grupo tratado con SIMPONI® IV y en el grupo tratado con el placebo fue 6.4. La disminución de la mediana (mejoría) desde el estado basal en la puntuación del impacto de la enfermedad en la productividad diaria (VAS) en los grupos con SIMPONI® IV y el placebo en la semana 14 y la semana 24 fue de -3.4 frente al -0.45 (p<0.001) y -3.3 frente al -0.6 (p<0.001), respectivamente.
Espondilitis anquilosante:
Estudio de EA: La seguridad y eficacia de SIMPONI® se evaluaron en un estudio en EA (GO-RAISE), un estudio multicéntrico, aleatorizado, doble ciego y controlado con placebo (hasta la semana 24) donde se evaluó el tratamiento de SIMPONI® 50 mg o 100mg administrado como inyección subcutánea cada 4 semanas en 356 pacientes adultos con EA activa. Los pacientes incluidos en este estudio fueron varones y mujeres que tenían síntomas de enfermedad activa (definida como un índice BASDAI ≥ 4 y una escala VAS para el dolor de espalda total ≥ 4, cada uno en una escala de 0 a 10 cm) a pesar del tratamiento vigente o previo con fármacos antirreumáticos modificadores de la enfermedad (FAMES) o fármacos antiinflamatorios no esteroideos (AINEs) y que no habían sido tratados previamente con terapia anti-TNFa. Los pacientes con anquilosis completa de la columna vertebral a nivel cervical y lumbar fueron excluidos del estudio. Los datos de eficacia fueron recogidos y analizados hasta la semana 256.
Los pacientes fueron aleatorizados y asignados al grupo del placebo (N=78), SIMPONI® 50mg (N=138) y SIMPONI® 100mg (N=140). El criterio de evaluación principal fue la respuesta ASAS 20 en la semana 14. Los principales criterios de evaluación secundaria incluyeron la respuesta ASAS 20 en la semana 24, Índice de funcionalidad Bath de la Espondilitis Anquilosante (BASFI) en la semana 14 e Índice de Metrología de Bath de la Espondilitis Anquilosante (BASMI) en la semana 14.
Estudio por vía IV de EA: La eficacia y seguridad de SIMPONI® IV se evaluaron en un estudio clínico multicéntrico, aleatorizado, doble ciego, controlado con placebo (GO-ALIVE) en 208 adultos con espondilitis anquilosante activa y con respuesta inadecuada o intolerantes a los AINEs. Los pacientes tenían un diagnóstico de EA definida por al menos 3 meses de acuerdo con el criterio modificado de Nueva York. Los pacientes presentaron síntomas de enfermedad activa (Índice de Bath para la actividad de la enfermedad EA [BASDAI] ≥ 4, VAS total para dolor de espalda ≥ 4, en escalas de 0 a 10 cm (0 to 100 mm), y un nivel de PCR ≥ 0.3 mg/dL (3 mg/L)). Los pacientes fueron aleatorizados para recibir SIMPONI® IV 2 mg/kg (N=105) o el placebo (N=103) como una infusión intravenosa de 30 minutos en la semana 0, 4 y 12. Todos los pacientes tratados con placebo recibieron SIMPONI® IV en la semana 16, semana 20 y posteriormente cada 8 semanas hasta la semana 52. Los pacientes en el grupo de tratamiento con SIMPONI® IV continuaron recibiendo las infusiones de SIMPONI® IV en la semana 20 y posteriormente cada 8 semanas hasta la Semana 52. Se permitió a los pacientes continuar con dosis estables concomitantes de MTX, SSZ, hidroxicloroquina (HCQ), corticoides orales a dosis baja (equivalente a ≤ 10 mg de prednisona por día), y/o AINEs durante el estudio. Durante el enrolamiento al estudio, se prohibió el uso de otros FAMEs, incluyendo agentes citotóxicos u otros agentes biológicos.
La mediana de la duración de la enfermedad EA fue 2.8 años, la mediana de la duración del dolor de espalda inflamatorio fue 8 años, el 90% de los pacientes fueron HLA-B27 positivos, el 8.2% había sido sometido a cirugía o a un procedimiento articular previo, el 5.8% presentaba anquilosis completa de la columna vertebral, el 14% había recibido terapia previa con un bloqueador de TNF (diferente de golimumab) y discontinuado por razones diferentes a la falta de eficacia dentro de las primeras 16 semanas del tratamiento (falla primaria), y el 76% había recibido por lo menos un FAME en el pasado. Durante el estudio, el uso de medicamentos concomitantes incluyó AINEs (88%), SSZ (38%), corticosteroides (26%), MTX (18%) y HCQ (0.5%).
El criterio de evaluación primario fue el porcentaje de los pacientes que alcanzaron una respuesta 20 en la evaluación en espondilitis anquilosante (ASAS) en la semana 16. Los principales criterios de evaluación secundarios fueron la proporción de sujetos que alcanzaron una respuesta ASAS 40 en la semana 16, la proporción de sujetos que alcanzaron una mejoría de por lo menos el 50% desde el estado basal en BASDAI en la semana 16 y el cambio desde el estado basal en BASFI en la semana 16.
Reducción de signos y síntomas
Estudio de EA: En general, no se observaron diferencias clínicamente significativas en la medida de la eficacia entre los regímenes de dosificación de SIMPONI® 50 y 100 mg en el estudio en EA de fase 3 hasta la semana 24. Según el diseño del estudio, a los pacientes en la extensión de largo plazo se les podia cambiar entre la dosis de 50 mg y 100 mg de SIMPONI® a discreción del médico del estudio.
El tratamiento con SIMPONI® causó un mejoría de los signos y síntomas según lo demostrado con la respuesta ASAS 20 en la semana 14. Un porcentaje significativamente mayor de los pacientes alcanzaron una respuesta ASAS 20 en el grupo combinado de SIMPONI® (50 mg y 100 mg de SIMPONI® juntos) y en los grupos individuales de la dosis de SIMPONI® en comparación con el placebo (p < 0.001 para todas las comparaciones) independientemente de los niveles de la PCR en la evaluación. El porcentaje de los pacientes que recibieron SIMPONI® y alcanzaron respuesta ASAS 20 fue 59% en comparación con el 22% en el grupo del placebo (Tabla 46). Se observó una mejoria en la primera evaluación (semana 4) después de la primera administración de SIMPONI® y se mantuvo hasta la semana 24 (Figura 8 y Tabla 46).
Figura 8: Estudio de EA: Porcentaje de pacientes con EA que alcanzaron respuesta ASAS 20 hasta la semana 24; pacientes aleatorizados en grupos de dosis del placebo y SIMPONI® 50 mg.
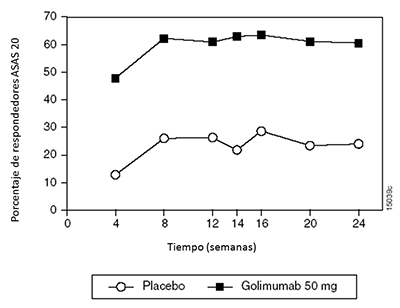
La respuesta ASAS 40, el cual representa por lo menos un 40% de la mejoría en el criterio de respuesta ASAS, se alcanzó en la semana 14 por más pacientes en el grupo de dosis de SIMPONI® 50 mg (45%) en comparación con el grupo del placebo (15%; p<0.001). La respuesta ASAS 40 se mantuvo hasta la semana 24 (Tabla 46).
Entre los pacientes que permanecieron en el estudio y tratados con SIMPONI®, la proporción de pacientes con una respuesta ASAS 20 y ASAS 40 fue similar desde la semana 24 hasta la semana 256.
En la semana 14, se alcanzó una actividad menor de la enfermedad (es decir, remisión parcial ASAS, definido como un valor < 2 en una escala de 0-10 cm en cada uno de los cuatro parámetros de la respuesta ASAS 20) del 23% en el grupo de SIMPONI® 50 mg frente al 5% en el placebo (p<0.001).
En comparación con el placebo, los pacientes tratados con SIMPONI® 50 alcanzaron una respuesta significativamente mayor medida por la respuesta ASAS 5/6 y un cambio del 50%, 70% y 90% desde el estado basal en el índice BASDAI (Tabla 46).
|
Tabla 46: Estudio de EA: Porcentaje de pacientes con EA con respuesta ASAS y BASDAI en pacientes aleatorizados |
||
|
Placebo (N=78)a |
SIMPONI® 50 mg* (N=138)a |
|
|
ASAS 20 (% de respondedores) |
||
|
Semana 14 |
22% |
59% |
|
Semana 24 |
23% |
56% |
|
ASAS 20 (% de respondedores) |
||
|
Semana 14 |
15% |
45% |
|
Semana 24 |
15% |
44% |
|
Remisión parcial ASAS (% de respondedores) |
||
|
Semana 14 |
5% |
23% |
|
Semana 24 |
5% |
27% |
|
ASAS 5/6 (% de respondedores) |
||
|
Semana 14 |
8% |
50% |
|
Semana 24 |
13% |
49% |
|
BASDAI 50 (% de respondedores) |
||
|
Semana 14 |
15% |
46% |
|
Semana 24 |
15% |
51% |
|
BASDAI 70 (% de respondedores) |
||
|
Semana 14 |
5% |
29% |
|
Semana 24 |
8% |
30% |
|
BASDAI 90 (% de respondedores) |
||
|
Semana 14 |
1% |
10% |
|
Semana 24 |
1% |
15% |
|
*p ≤ 0.00a para todas las comparaciones, excepto en BASDAI 90 en la semana 14, donde p = 0.017. a N refleja los pacientes aleatorizados; el número real de los pacientes evaluables para cada criterio de evaluación puede variar con el punto cronológico. Una respuesta ASAS 20 (Anderson et al, 2001) fue definido como: 1. Una mejoría ≥ 20% desde el estado basal y una mejoría absoluta desde el estado basal de por lo menos 1 en una escala de 0 a 10 cm en por lo menos 3 de los siguientes 4 parámetros: evaluación general del paciente, evaluación del dolor (dolor de espalda total), puntuación BASFI o inflamación (promedio de las primeras 2 preguntas del índice BASDAI en relación con la rigidez matutina). 2. Ausencia de deterioro desde el estado basal (el deterioro se define como ≥ 20% de empeoramiento y empeoramiento específico de por lo menos 1 en una escala de 0 a 10 cm) en el parámetro potencial restante. ASAS 40: Un ASAS 40 se define como ≥ 40% de mejoría en 3 de los 4 parámetros, con una mejoría absoluta de por lo menos 2 en una escala de 0 a 10 cm y sin deterioro en el parámetro restante. Remisión ASAS parcial: Cada uno de los 4 parámetros del ASAS 20 tiene una puntuación de menos de 2. ASAS 5/6 se define como 20% o más de mejoría en cualquiera de los 5 de los 6 parámetros del dolor total de espalda en la escala VAS, paciente en general en la escala VAS, función (BASFI), rigidez matutina (promedio de por lo menos 2 preguntas del índice BASDAI), proteína C-reactiva y medida de la flexión lateral lumbar. BASDAI: Autoevaluación del paciente usando una escala VAS (0 a 10 cm) de los siguientes criterios: Fatiga, dolor de columna, dolor de la articulación, entésitis, rigidez cualitativa matutina y rigidez cuantitativa matutina. |
||
Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, tasas similares del cambio desde el estado basal en el índice BASDAI se observaron desde la semana 24 hasta la semana 256.
Los pacientes tratados con SIMPONI® 50 mg alcanzaron una mejoría significativamente en todos los componentes del ASAS 20 en comparación con el placebo (Tabla 47 y Tabla 51).
|
Tabla 47: Estudio en EA: Mejoría en los componentes del ASAS en pacientes aleatorizados |
||
|
Placebo (N = 78)a |
SIMPONI® 50mg* (N = 138)a |
|
|
Evaluación general del paciente de la actividad de la enfermedadb: (mediana del cambio desde el estado basal) |
||
|
Estado basal (mediana) |
7.2 |
7.0 |
|
Semana 14 |
-0.8 |
-2.8 |
|
Semana 24 |
-0.2 |
-2.6 |
|
Dolor de espalda totalb: (mediana del cambio desde el estado basal) |
||
|
Estado basal (mediana) |
7.6 |
7.5 |
|
Semana 14 |
-0.8 |
-3.5 |
|
Semana 24 |
-0.4 |
-3.5 |
|
Inflamaciónc: (mediana del cambio desde el estado basal en rigidez matutina) |
||
|
Estado basal (mediana) |
7.1 |
7.1 |
|
Semana 14 |
-0.5 |
-3.2 |
|
Semana 24 |
-0.2 |
-3.6 |
|
Dolor de espalda nocturnab: (mediana del cambio desde el estado basal) |
||
|
Estado basal (mediana) |
7.4 |
7.1 |
|
Semana 14 |
-0.3 |
-3.0 |
|
Semana 24 |
-0.4 |
-3.1 |
|
Proteína C-reactivad: (mediana del cambio desde el estado basal en mg/dL) |
||
|
Estado basal (mediana) |
1.2 |
1.1 |
|
Semana 14 |
0 |
-0.7 |
|
Semana 24 |
0 |
-0.7 |
|
*p<0.001 para todas las comparaciones a N refleja los pacientes aleatorizados; el número real de los pacientes evaluables para cada criterio de evaluación varia en el punto cronológico. b Escala Analógica Visual (con 0 = “mejor” y 10 = “peor”. Una puntuación negativa/decreciente es indicativo de mejoría. c Promedio de por lo menos 2 preguntas en las 6 preguntas en el índice BASDAI. d Rango normal 0 -0.6mg/dL. |
||
Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, tasas similares de la mejoría en los componentes medidos del ASAS 20 (evaluación general del paciente de la actividad de la enfermedad, dolor de espalda total y dolor de espalda nocturna) se observaron desde la semana 24 hasta la semana 256.
Estudio por vía IV de EA: El tratamiento con SIMPONI® IV, en comparación con el placebo, produjo una mejoría significativa en los signos y síntomas según lo demostrado por el porcentaje de los pacientes con una respuesta ASAS 20 en la semana 16 (ver Tabla 48). En los pacientes tratados con SIMPONI® IV, el 73% de los pacientes alcanzó respuesta ASAS 20 en comparación con el 26% tratados con el placebo (p<0.001) en la semana 16. Además, se alcanzó respuesta ASAS 40 en la semana 16 por un mayor número de pacientes tratados con SIMPONI® IV (48%) en comparación con el grupo del placebo (8.7%; p<0.001). En la semana 16, un mayor porcentaje de los pacientes tratados con SIMPONI® IV alcanzó un nivel bajo de la actividad de la enfermedad (es decir, remisión parcial ASAS, definida como un valor < 2 en una escala de 0-10 cm en cada uno de los 4 parámetros de respuesta ASAS 20) en comparación con los pacientes tratados con el placebo (16% frente al 3.9%, p=0.003). En comparación con el placebo, los pacientes tratados con SIMPONI® IV alcanzaron respuestas significativamente mayores medidas por la respuesta ASAS 5/6 (65% frente al 12%, respectivamente, p<0.001).
|
Tabla 48: Estudio por vía IV de EA: Porcentaje de respondedores ASAS en la semana 16: pacientes aleatorizados |
|||
|
Placebo Na=103 |
SIMPONI® IV Na=105 |
Diferencia de los tratamientos (IC del 95%) |
|
|
% de respondedores |
|||
|
ASAS 20 |
26% |
73% |
47%b (35.2, 59.0) |
|
ASAS 40 |
8.7% |
48% |
39%b (27.9, 49.8) |
|
Remisión parcial ASAS |
3.9% |
16% |
12.3%c (4.4, 20.3) |
|
ASAS 5/6 |
12% |
65% |
53.0%b (42.1, 64.0) |
|
a N refleja a los pacientes aleatorizados. El texto en negrita indica el criterio de evaluación primario. b p<0.001 c p=0.003 |
|||
El porcentaje de los pacientes que alcanzaron respuestas ASAS 20 por visita hasta la semana 16 para el Estudio por vía IV de EA se muestra en la Figura 9. Se observaron respuestas ASAS 20 en el 37% de los pacientes tratados con SIMPONI® IV en la primera evaluación (semana 2) en comparación con el 19% de los pacientes tratados con el placebo.
Figura 9: Estudio por vía IV de EA: Porcentaje de pacientes que alcanzaron una respuesta ASAS 20 hasta la semana 16: pacientes aleatorizados
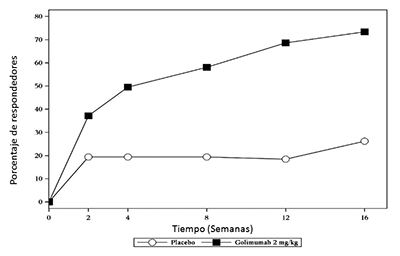
La Tabla 49 muestra las mejorías en los componentes del criterio de respuesta ASAS y otras medidas de la actividad de la enfermedad en la semana 16 para los grupos con SIMPONI® IV y placebo.
|
Tabla 49: Estudio por vía IV de EA: Cambio desde el estado basal en los componentes ASAS y otras medidas de la actividad de la enfermedad en la semana 16: pacientes aleatorizados |
||||
|
Placebo N=103a |
SIMPONI® IV N=105a |
|||
|
Estado basal |
Cambio en la semana 16 desde el estado basal |
Estado basal |
Cambio en la semana 16 desde el estado basal |
|
|
Criterio de respuesta ASAS 20 |
||||
|
Evaluación global del pacientes de la actividad de la enfermedad (0-100 mm)b |
73 |
-4.0 |
73 |
-30 |
|
Dolor total de espalda 0-100 mm)c |
74 |
-6.0 |
72 |
-32 |
|
BASFI (0-10)d |
6.6 |
-0.3 |
6.5 |
-2.2* |
|
Inflamación (0-10)e |
7.5 |
-0.8 |
7.1 |
-3.3 |
|
Dolor de espalda por la noche (0-100 mm)c |
73 |
-3.5 |
73 |
-33 |
|
PCR (mg/L) |
15 |
-1.4 |
15 |
-13 |
|
Nota: Todos los valores son medianas. * p<0.001 a N refleja a los pacientes aleatorizados; los números reales de pacientes evaluables para cada criterio de evaluación pueden variar. b Medido en una escala analógica visual (VAS) donde 0 = muy bien, 100 = muy mal c Medido en una escala analógica visual (VAS) donde 0 = ausencia de dolor, 100 = dolor extremadamente severo d BASFI es el Índice de funcionalidad de Bath de la espondilitis anquilosante. e Inflamación es el promedio de 2 autoevaluaciones de rigidez matutina en el BASDAI. |
||||
BASDAI es una autoevaluación del paciente utilizando una escala analógica visual (0 a 10 cm) con los siguientes criterios: fatiga, dolor de columna, dolor de articulaciones, entesitis, rigidez matutina cualitativa y rigidez matutina cuantitativa. Los pacientes tratados con SIMPONI® IV alcanzaron respuestas significativamente mayores medidas por una mejoría del 50%, 70% y 90% desde el estado basal en BASDAI en la semana 16 (Tabla 50). La mediana de la puntuación BASDAI en el estado basal, evaluada en una escala VAS (0-10 cm), fue 7.1 para el grupo tratado con SIMPONI® IV y 7.0 para el grupo del placebo. La disminución de la mediana (mejoría) desde el estado basal en la puntuación BASDAI fue mayor en los pacientes tratados con SIMPONI® IV en comparación con los pacientes tratados con el placebo en la semana 16 (-3.0 frente al -0.7, respectivamente). El porcentaje de los pacientes que alcanzaron una respuesta BASDAI 50 en la semana 16 fue 41% para los pacientes tratados con SIMPONI® IV y 15% para los pacientes tratados con el placebo (p<0.001). En la semana 16, una mayor proporción de los pacientes tratados con SIMPONI® IV alcanzó una puntuación BASDAI menor de 3 en comparación con los pacientes tratados con el placebo (38% frente al 13%, respectivamente).
|
Tabla 50: Estudio por vía IV de EA: Porcentaje de pacientes con respuesta BASDAI en la semana 16, pacientes aleatorizados |
|||
|
Placebo Na=103 |
SIMPONI® IV Na=105 |
Diferencia de los tratamientos (IC del 95%) |
|
|
Respondedores, % de los pacientes |
|||
|
BASDAI 50 |
15% |
41% |
26.4* (14.8, 37.9) |
|
BASDAI 70b |
5.8% |
25% |
19.0 (9.6, 28.4) |
|
BASDAI 90b |
2.9% |
11% |
8.5 (1.6, 15.3) |
|
Puntuación BASDAI <3b |
13% |
38% |
25.5 (14.2, 36.7) |
|
* p<0.001 a N refleja a los pacientes aleatorizados b BASDAI: Autoevaluación del paciente utilizando una VAS (0 a 10 cm) con los siguientes criterios: fatiga, dolor de columna, dolor de articulaciones, entesitis, rigidez matutina cualitativa y rigidez matutina cuantitativa. BASDAI 50, BASDAI 70 y BASDAI 90 son las proporciones de sujetos que alcanzaron una mejoría de ≥ 50%, ≥ 70% o ≥ 90% desde el estado basal en la puntuación BASDAI. |
|||
Los pacientes con entesitis en el estado basal (83%) fueron evaluados para determinar la mejoría en la entesitis usando el Índice de entesitis UCSF en una escala de 0-17. En el estado basal, la mediana de la puntuación del Índice de entesitis UCSF en el grupo tratado con SIMPONI® IV y en el grupo tratado con el placebo fue 5.0 y 6.0, respectivamente. Los pacientes tratados con SIMPONI® IV mostraron una mejoría en la puntuación de entesitis de 3.0 en comparación con una mejoría en los pacientes tratados con el placebo de 1.0 en la semana 16 (p<0.001).
La puntuación de la Actividad de la Enfermedad Espondilitis Anquilosante (ASDAS) es una medida de la actividad de la enfermedad y se deriva de la evaluación del dolor total de espalda, rigidez matutina, evaluación global del paciente, dolor/inflamación periférica y medición de la PCR. Una mejoría importante en ASDAS se define como una disminución ≥ 2.0 y una enfermedad inactiva se define como una puntuación de <1.3. En los pacientes tratados con SIMPONI® IV, el 59% de los pacientes alcanzó mejoría importante ASDAS en comparación con el 2.9% de los pacientes tratados con el placebo (p<0.001) en la semana 16. En la semana 16, un mayor porcentaje de los pacientes tratados con SIMPONI® IV alcanzó una enfermedad inactiva en comparación con los pacientes tratados con el placebo (28% frente al 2.9%, p<0.001).
Mejoría de la función física:
Estudio de EA: El tratamiento con SIMPONI® causó una mejoría significativa de la función física evaluada mediante el cambio del índice BASFI desde el estado basal y en el porcentaje de los pacientes que alcanzaron una mejoría de 2 unidades del índice BASFI en la semana 14 y 24. La mediana de la mejoría del índice BASFI en la semana 14 fue 1.4 en el grupo de SIMPONI® 50mg en comparación con el empeoramiento de 0.1 en el grupo del placebo (p<0.001). La mejoría de la función física se mantuvo en la semana 24 en el grupo de pacientes tratados con SIMPONI® (Tabla 51). Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, se observaron tasas similares de mejoría de la función física desde la semana 24 hasta la semana 256.
|
Tabla 51: Estudio de EA: Análisis de la mejoría del índice BASFI en pacientes aleatorizados |
||
|
Placebo |
SIMPONI® 50 mg * |
|
|
BASFI (0-10): mediana del cambio desde el estado basal |
||
|
Estado basal (mediana) |
4.9 |
5.0 |
|
Semana 14 |
0.1 |
-1.4 |
|
Semana 24 |
0.4 |
-1.6 |
|
Mejoría > 2 unidades del BASFI desde el estado basal: % de pacientes |
||
|
Semana 14 |
17% |
48% |
|
Semana 24 |
14% |
50% |
|
* p<0.001 para todas las comparaciones a N representa los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación varía en el punto cronológico. El índice de funcionalidad de Bath de la Espondilitis Anquilosante (BASFI), promedio de 10 preguntas. Una puntuación negativa/decreciente es indicativo de la mejoría del BASFI. |
||
Estudio por vía IV de EA: El Índice de funcionalidad de Bath de la espondilitis anquilosante (BASFI) es el promedio de 10 preguntas de autoevaluación relacionadas con la anatomía funcional del paciente y la capacidad para sobrellevar la vida cotidiana. Una disminución de la puntuación indica mejoría en BASFI. La mediana de la puntuación BASFI en el estado basal fue 6.3 y 6.1 para los grupos con SIMPONI® IV y el placebo, respectivamente. El tratamiento con SIMPONI® IV produjo mejorías significativas en la función física evaluada por los cambios desde el estado basal en BASFI a través del tiempo y el porcentaje de los pacientes que alcanzaron una mejoría de 2 unidades en BASFI en la semana 16. En la semana 2 y 16, la disminución de la mediana respectiva (mejoría) desde el estado basal en BASFI para los grupos con golimumab y el placebo fue -0.9 y -0.3 en la semana 2 y de -2.2 frente al -0.3 en la semana 16. La mejoría en la función física se mantuvo en la semana 28 en los pacientes tratados con SIMPONI® IV. Entre los pacientes con una puntuación BASFI ≥ 2 en el estado basal, un mayor porcentaje de los pacientes tratados con SIMPONI® IV alcanzó una mejoría de 2 unidades en BASFI en comparación con los pacientes tratados con el placebo (59% frente al 22%, respectivamente) en la semana 16 (ver Tabla 52).
|
Tabla 52: Estudio por vía IV de EA: Cambio desde el estado basal en los análisis de BASFI; pacientes aleatorizados |
||
|
Placebo N=103a |
SIMPONI® IV N=105a |
|
|
BASFI (0-10): cambio de la mediana desde el estado basalb |
||
|
Estado basal (mediana) |
6.6 |
6.5 |
|
Semana 16 |
-0.3 |
-2.2 |
|
Mejoría > 2 unidades en BASFI desde el estado basalb: % de pacientes |
||
|
Semana 16 |
22% |
59% |
|
a N refleja a los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar por tiempo de evaluación. b Índice de funcionalidad de Bath de la espondilitis anquilosante (BASFI), promedio de 10 preguntas de autoevaluación relacionadas con la anatomía funcional del paciente y la capacidad para sobrellevar la vida cotidiana. Una puntuación negativa/decreciente indica mejoría en BASFI. |
||
Mejoría en el rango de movimiento:
Estudio de EA: Cambios no significativos en BASMI se observó en la semana 14 o en la semana 24 en el grupo de SIMPONI® 50 mg en comparación con el grupo del placebo. Sin embargo, el porcentaje de pacientes por lo menos con 1-unidad de mejoria en BASMI en la semana 14 en el grupo de SIMPONI® 50 mg fue significativamente mayor en comparación con el grupo del placebo. Cada uno de los 5 componentes de BASMI se analizó en la semana 14 y en la semana 24. En el grupo de SIMPONI® 50 mg frente al placebo, una mediana significante de la mejoria desde el estado basal se observó en la semana 14 y en la semana 24 para la flexión lumbar, para la flexión lateral lumbar en la semana 24, y la medida de la distancia intermaleolar en la semana 14 y en la semana 24. El incremento de la mediana de la expansión torácica en la semana 24 fue significativo para SIMPONI® 50 mg frente al placebo (p=0.013) (Tabla 53). Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, tasas similares de la mejoría en BASMI se observaron desde la semana 24 hasta la semana 256.
|
Tabla 53: Estudio de EA: Análisis de la movilidad de la columna en pacientes aleatorizados |
|||
|
Placebo (N = 78)a |
SIMPONI® 50 mg (N = 138)a |
Valor-p |
|
|
BASMI (0-10): Cambio de la mediana desde el estado basal |
|||
|
Estado basal (mediana) |
4.0 |
3.0 |
|
|
Semana 14 |
0 |
0 |
NS |
|
Semana 24 |
0 |
0 |
NS |
|
Mejoria ≥ 1-unidad en BASMI desde el estado basal: % de pacientes |
|||
|
Semana 14 |
29% |
44% |
0.037 |
|
Semana 24 |
34% |
47% |
NS |
|
Flexión lumbar (Prueba de Schober modificado) (cm): Cambio de la mediana desde el estado basal |
|||
|
Estado basal (mediana) |
3.0 |
3.4 |
|
|
Semana 14 |
0 |
0.2 |
0.006 |
|
Semana 24 |
0 |
0.5 |
0.040 |
|
Flexión lateral lumbar (cm): Cambio de la mediana desde el estado basal |
|||
|
Estado basal (mediana) |
9.5 |
11.5 |
|
|
Semana 14 |
0.5 |
1.0 |
NS |
|
Semana 24 |
1.0 |
2.0 |
0.012 |
|
Distancia intermaleolar (cm): Cambio de la mediana desde el estado basal |
|||
|
Estado basal (mediana) |
100.0 |
103.5 |
|
|
Semana 14 |
-1.3 |
2.5 |
0.016 |
|
Semana 24 |
-2.0 |
6.0 |
0.001 |
|
Distancia del trago de la oreja a la pared (cm): Cambio de la mediana desde el estado basal |
|||
|
Estado basal (mediana) |
13.0 |
12.0 |
|
|
Semana 14 |
0 |
0 |
NS |
|
Semana 24 |
0 |
0 |
NS |
|
Rotación cervical (°): Cambio de la mediana desde el estado basal |
|||
|
Estado basal (mediana) |
47.0 |
50.0 |
|
|
Semana 14 |
1.0 |
4.0 |
NS |
|
Semana 24 |
4.0 |
5.5 |
NS |
|
Expansión torácica (cm): Cambio de la mediana desde el estado basal |
|||
|
Estado basal (mediana) |
3.50 |
3.50 |
|
|
Semana 14 |
0 |
0 |
NS |
|
Semana 24 |
0 |
0.5 |
0.013 |
|
aN refleja a los pacientes aleatorizados, número real de los pacientes evaluables para cada criterio de evaluación podria variar en el punto cronológico. NS = No significativo Valores normales de la movilidad de la columna: Prueba de Schober modificado: > 4 cm; flexión lateral lumbar: > 10 cm; distancia intermaleolar: > 100 cm; distancia del trago de la oreja a la pared: < 15 cm; rotación cervical: > 70° cm; expansión torácica: >6.5 cm en varones, > 4.5 cm en mujeres, de 35 – 45 años. |
|||
Estudio por vía IV de EA: El Índice de metrología de Bath de la espondilitis Anquilosante (BASMI) es una evaluación musculoesquelética y se representa como una puntuación combinada de 5 componentes (flexión lumbar, flexión lateral lumbar, distancia intermaleolar, distancia del trago de la oreja a la pared y rotación cervical). En la semana 16, la disminución de la mediana (mejoría) desde el estado basal en BASMI fue de mayor magnitud en los pacientes tratados con SIMPONI® IV (-0.4) en comparación con el placebo (-0.05, p=0.001). El porcentaje de los pacientes con una mejoría de por lo menos 1 unidad en BASMI en la semana 16 en los pacientes tratados con SIMPONI® IV (14%) fue mayor en comparación con los pacientes tratados con el placebo (5.0%, p=0.029) (ver Tabla 54).
|
Tabla 54: Estudio por vía IV de EA: Cambio de la mediana desde el estado basal en los componentes de la puntuación BASMI y otras medidas de la movilidad de la columna; pacientes aleatorizados |
|||
|
Placebo (N=103)a |
SIMPONI® IV (N=105)a |
Valor de p |
|
|
BASMI (0-10): cambio de la mediana desde el estado basal |
|||
|
Estado basal (mediana) |
5.0 |
5.0 |
|
|
Semana 16 |
-0.1 |
-0.4 |
<0.001 |
|
Mejoria ≥ 1 unidad en BASMI desde el estado basal: % de pacientes |
|||
|
Semana 16 |
5.0% |
14.0% |
0.029 |
|
Flexión lumbar (Prueba de Schober modificado): cambio de la mediana desde el estado basalb |
|||
|
Estado basal (mediana) |
6.3 |
6.3 |
|
|
Semana 16 |
0.0 |
-0.4 |
0.026 |
|
Flexión lateral lumbar: cambio de la mediana desde el estado basalb |
|||
|
Estado basal (mediana) |
6.0 |
6.1 |
|
|
Semana 16 |
0.00 |
-0.5 |
<0.001 |
|
Distancia intermaleolar: cambio de la mediana desde el estado basalb |
|||
|
Estado basal (mediana) |
3.5 |
3.5 |
|
|
Semana 16 |
-0.3 |
-0.6 |
0.047 |
|
Distancia del trago de la oreja a la pared (cm): Cambio de la mediana desde el estado basalb |
|||
|
Estado basal (mediana) |
0.2 |
0.1 |
|
|
Semana 16 |
0.0 |
0.0 |
NS |
|
Rotación cervical: Cambio de la mediana desde el estado basalb |
|||
|
Estado basal (mediana) |
8.8 |
8.8 |
|
|
Semana 16 |
0.0 |
0.0 |
NS |
|
Expansión torácica (cm): Cambio de la mediana desde el estado basal |
|||
|
Estado basal (mediana) |
3.0 |
3.0 |
|
|
Semana 16 |
0.0 |
0.8 |
NS |
|
a N refleja a los pacientes aleatorizados; el número real de los pacientes evaluables para cada criterio de evaluacipon puede variar por punto de evaluación. b Los valores de los componentes de BASMI se presentan como puntuaciones S, usando el cálculo de van der Heijde. |
|||
Mejoría de la calidad de vida relacionada con la salud:
Estudio de EA: En el estudio en EA, los pacientes tratados con SIMPONI® 50 mg mostraron una media de la mejoría significativamente mayor desde el estado basal de la puntuación del PCS del SF-36 en comparación con el placebo en la semana 14 (7.3 frente a 2.4; p<0.001) y en la semana 24 (7.9 frente a 2.0; p<0.001). El promedio en el estado basal de la puntuación del PCS de 29.3 ± 8.3 y 29.2 ± 7.1 en el grupo de SIMPONI® y en el grupo del placebo, respectivamente, fue menor que la norma de 50 ± 10 medido en la población de Estados Unidos. El cambio promedio desde el estado basal de la puntuación del PCS de SF-36 en la semana 14 fue mayor en el grupo de SIMPONI® 50 mg que en el grupo del placebo (8.8 ± 9.6 frente a 3.0 ± 7.2, respectivamente). El promedio de la mejoría de la función física del PCS del SF-36 se mantuvo hasta la semana 24 (Tabla 55). Entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI®, tasas similares de la media de la mejoria de la función física del PCS del SF-36 se observaron desde la semana 24 hasta la semana 256.
Los pacientes tratados con SIMPONI® 50 mg mostraron una mediana de la mejoria significativamente mayor desde el estado basal del MCS del SF-36 en comparación con el placebo en la semna 14 (1.5 frente a 0.1; p = 0.014). La media del cambio desde el estado basal de la puntuación del MCS del SF-36 en la semana 14 fue mayor en el grupo de SIMPONI® 50 mg que en el grupo del placebo (2.6 ± 8.5 frente a -0.4 ± 8.3, respectivamente). La mejoría de la función mental del MCS del SF-36 no se mantuvo hasta la semana 24 (Tabla 55).
|
Tabla 55: Estudio de EA: Análisis del SF-36 en pacientes aleatorizados |
|||
|
Placebo (N = 78)a |
SIMPONI® 50 mg (N = 138)a |
Valor-p |
|
|
PCS SF-36 |
|||
|
Estado basal |
|||
|
Media ± S.D. |
29.2 ± 7.1 |
29.3 ± 8.3 |
|
|
Mediana |
28.3 |
29.7 |
|
|
Semana 14, mejoría desde el estado basal |
|||
|
Media ± S.D. |
3.0 ± 7.2 |
8.8 ± 9.6 |
|
|
Mediana |
2.40 |
7.30 |
< 0.001 |
|
Semana 24, mejoría desde el estado basal |
|||
|
Media ± S.D. |
2.8 ± 8.1 |
9.6 ± 10.6 |
|
|
Mediana |
2.0 |
7.9 |
<0.001 |
|
MCS SF-36 |
|||
|
Estado basal |
45.7 ± 10.7 |
45.1 ± 10.5 |
|
|
Media ± S.D. |
46.2 |
46.5 |
|
|
Mediana |
|||
|
Semana 14, mejoría desde el estado basal |
|||
|
Media ± S.D. |
-0.4 ± 8.3 |
2.6 ± 8.5 |
|
|
Mediana |
0.1 |
1.5 |
0.014 |
|
Semana 24, mejoría desde el estado basal |
|||
|
Media ± S.D. |
0.7 ± 9.5 |
2.3 ± 8.3 |
|
|
Mediana |
-0.3 |
1.4 |
0.283 |
|
a N refleja los pacientes aleatorizados; el número real de los pacientes evaluables para cada criterio de evaluación varía en el punto cronológico. El valor promedio de la puntuación del componente físico (PCS) y la puntuación del componente mental (MCS) del SF-36 es 50 ± 10 cada uno para la población de Estados Unidos. El rango es de 0 (peor) a 100 (mejor). |
|||
Estudio por IV de EA: La calidad de vida relacionada con la salud se midió utilizando los resúmenes de los componentes físicos y mentales de SF-36 (Encuesta de salud 36 forma abreviada), ASQoL (cuestionario de calidad de vida para espondilitis anquilosante), la puntuación del Índice EQ 5D-5L (cuestionario de 5 dimensiones EuroQoL) y la puntuación EQ VAS en el estado basal, semana 8, semana 16 y semana 28.
Los pacientes que recibieron SIMPONI® IV demostraron mejoría significativamente mayor de la mediana desde el estado basal en comparación con el placebo en la puntuación del resumen del componente físico (PCS, 7.8 frente al 2.7, p<0.001) en la semana 16. En el estado basal, la mediana de las puntuaciones del PCS del SF-36 fueron 31 en el grupo con SIMPONI® IV y 31 para el grupo del placebo. Mayores proporciones de los sujetos en el grupo con SIMPONI® IV en comparación con el placebo alcanzaron un cambio clínicamente significativo desde el estado basal en la puntuación del PCS del SF-36 (un incremento de 5 o más unidades) en la semana 8 (58% frente al 27%) y en la semana 16 (68% frente al 36%).
Los pacientes que recibieron SIMPONI® IV demostraron mejoría significativamente mayor de la mediana desde el estado basal en comparación con el placebo en la puntuación del resumen del componente mental (MCS, 5.7 frente al 1.8, p<0.001) en la semana 16. En el estado basal, la mediana de las puntuaciones del MCS del SF-36 fue 40 en el grupo de SIMPONI® IV y 41 para el grupo de placebo. Mayores proporciones de los sujetos en el grupo de SIMPONI® IV en comparación con el placebo alcanzaron un cambio clínicamente significativo desde el estado basal en la puntuación del MCS del SF-36 (un incremento de 5 o más unidades) desde la semana 8 (49% frente al 34%) y en la semana 16 (54% frente al 29%).
Además, las ocho áreas del SF-36 que comprenden las puntuaciones del PCS y MCS mostraron mayores mejorias en la mediana en la semana 8 y la semana 16 para los pacientes tratados con SIMPONI® IV en comparación con el placebo.
El Cuestionario de calidad de vida de espondilitis anquilosante (ASQoL) es un instrumento específico para la enfermedad utilizado para medir la calidad de vida en la población de pacientes con EA. Consta de 18 áreas que requieren una respuesta de sí o no a preguntas con el impacto del dolor en el sueño, estado de ánimo, motivación, habilidad para sobrellevar la vida cotidiana, actividades cotidianas, independencia, relaciones y vida social. Puntuaciones bajas indican mejoría. La mediana de las puntuaciones de ASQoL en el estado basal en el grupo tratado con SIMPONI® IV y en el grupo con el placebo fue 14 y de 13, respectivamente. Los pacientes tratados con SIMPONI® IV alcanzaron una mayor disminución de la mediana (mejoría) desde el estado basal en la puntuación ASQoL en comparación con los pacientes tratados con el placebo en la semana 16 (-4.0 frente al -1.0, p<0.001).
EuroQoL 5D-5L es una medida del estado de salud para proporcionar una medida de la salud en evaluaciones clínicas y económicas. EQ-5D-5L consta de 2 elementos: el sistema descriptivo EQ-5D-5L y la escala analógica visual EQ (EQ VAS). El sistema descriptivo EQ-5D incluye las 5 áreas siguientes: movilidad, autocuidado, actividades usuales, dolor/malestar y ansiedad/depresión. El EQ VAS registra la salud autocalificada por el respondedor en una VAS con los criterios de evaluación “la peor salud imaginable” y “la mejor salud imaginable”.
Los pacientes tratados con SIMPONI® IV alcanzaron mejorías en la mediana desde el estado basal en la puntuación del Índice EQ-5D-5L y en la puntuación EQ VAS en comparación con los pacientes tratados con el placebo en la semana 16 (0.2 frente al 0.05 y 20 mm frente al 5.0 mm, respectivamente). Los valores de la mediana en el estado basal para la puntuación del índice EQ-5D-5L y la puntuación EQ VAS en el grupo tratado con SIMPONI® IV y en el grupo del placebo fue 0.5 frente al 0.6 y de 37 mm frente al 37 mm, respectivamente (ver Tabla 56).
|
Tabla 56: Estudio por IV de EA: Análisis de la calidad de vida relacionada con la salud en la semana 16; pacientes aleatorizados y tratados |
||
|
Placebo (N=103)a |
SIMPONI® IV (N=105)a |
|
|
PCS del SF-36 |
||
|
Puntuación en el estado basal |
||
|
Media ± DE |
32 ± 5.9 |
32 ± 5.6 |
|
Mediana |
31 |
31 |
|
Semana 16, mejoría desde el estado basal |
||
|
Media ± DE |
2.9 ± 6.2) |
8.5 ± 7.5 |
|
Mediana |
2.7 |
7.8 |
|
MCS del SF 36 |
||
|
Puntuación en el estado basal |
||
|
Media ± DE |
42 ± 10.2 |
40 ± 10.4 |
|
Mediana |
41 |
40 |
|
Semana 16, mejoría desde el estado basal |
||
|
Media ± DE |
0.8 ± 10.0 |
6.5 ± 9.1 |
|
Mediana |
1.8 |
5.7 |
|
Puntuación ASQoL |
||
|
Puntuación en el estado basal |
||
|
Media ± DE |
12 ± 4.1 |
13 ± 4.0 |
|
Mediana |
13 |
14 |
|
Semana 16, mejoría desde el estado basalb |
||
|
Media ± DE |
-1.8 ± 4.6 |
-5.4 ± 5.0 |
|
Mediana |
-1.0 |
-4.0 |
|
Índice EQ 5Dc |
||
|
Puntuación en el estado basalc |
||
|
Media ± DE |
0.6 ± 0.15 |
0.5 ± 0.16 |
|
Mediana |
0.6 |
0.5 |
|
Semana 16, mejoría desde el estado basal |
||
|
Media ± DE |
0.05 ± 0.14 |
0.2 ± 0.16 |
|
Mediana |
0.05 |
0.2 |
|
EQ 5D VASc |
||
|
Puntuación en el estado basal (mm) |
||
|
Media ± DE |
41 ± 18.1 |
42 ± 22.7 |
|
Mediana |
37 |
37 |
|
Semana 16, (mm), mejoría desde el estado basal |
||
|
Media ± DE |
4.8 ± 23.4 |
20 ± 24.6 |
|
Mediana |
5.0 |
20 |
|
a N refleja a los pacientes aleatorizados; el número real de pacientes evaluables para cada criterio de evaluación puede variar por punto de evaluación. b Una puntuación negativa/decreciente indica mejoría. c EuroQoL 5D-5L es una medida del estado de salud para proporcionar una medida de la salud en evaluaciones clínicas y económicas. EQ-5D-5L incluye 2 elementos: el sistema descriptivo EQ-5D-5L y la escala analógica visual de EQ (EQ VAS). El sistema descriptivo EQ-5D incluye las siguientes 5 áreas: movilidad, autocuidado, actividades usuales, dolor/malestar y ansiedad/depresión. EQ VAS registra el estado de salud autocalificado por el respondedor en una VAS con los criterios de evaluacón “la peor salud imaginable” y “la mejor salud imaginable”. |
||
Economía sanitaria:
Estudio de EA: Los datos de economía sanitaria sobre utilización de recursos sanitarios, tiempo laboral perdido por los pacientes y cuidadores, empleabilidad y productividad del paciente en el trabajo, el colegio, o el hogar se recogieron por medio de cuestionarios aplicados en el estado basal y posteriormemnte cada 8 semanas. Se les solicitó a los pacientes que indicaran cuánto su enfermedad afectó su productividad en el trabajo, colegio u hogar en las 4 semanas previas usando una escala VAS de 0 a 10 cm (de no afectada a sumamente afectada). A continuación se presentan solamente aquellas evaluaciones en las cuales se observaron diferencias estadísticamente significativas entre SIMPONI® 50 mg y placebo.
En el estado basal, el grupo de SIMPONI® 50 mg y el grupo del placebo tuvieron puntuación comparable de la mediana de la productividad (6.8 frente a 6.6, respectivamente). La mediana de la mejoría en el autoreporte sobre la productividad fue significativamente mayor en el grupo de SIMPONI® 50 mg que en el grupo del placebo en la semana 16 (-2.5 frente a -0.2; p < 0.001) y en la semana 24 (-2.3 frente a -0.2; p < 0.001). La media de la mejoría en el autoreporte sobre la productividad en el grupo de SIMPONI® 50 mg y el grupo del placebo en la semana 16 y en la semana 24 fue de -2.8 ± 3.0 frente a -0.4 ± 2.7 y -2.7 ± 3.1 frente a -0.4 ± 3.0, respectivamente.
Estudio por via IV de EA: Se recogieron datos de economía de salud sobre la productividad de los pacientes en el trabajo a través del Cuestionario de limitaciones laborales (WLQ) y la escala analógica visual de productividad (VAS) en el estado basal y en las semanas 8, 16 y 28.
El WLQ es un cuestionario para evaluar la pérdida de productividad laboral relacionada con la salud. Se solicitó a los pacientes que trabajaban tiempo completo o medio tiempo o de manera voluntaria que calificaran su nivel de dificultad o la capacidad para realizar tareas laborales específicas a fin de evaluar la pérdida de productividad laboral relacionada con la salud a través del cuestionario en una escala de 0 (sin limitaciones en ningún momento) a 100 (limitado todo el tiempo). Posteriormente, las puntuaciones de la escala se convirtieron a una puntuación de pérdida de productividad que indica un porcentaje estimado de pérdida de productividad laboral por motivos de salud. En el estado basal, la mediana de las puntuaciones de la pérdida de productividad WLQ en el grupo tratado con SIMPONI® IV y en el grupo tratado con el placebo fue 11 y 11, respectivamente. La disminución de la mediana (mejoría) desde el estado basal en la puntuación de la pérdida de productividad WLQ en los grupos con SIMPONI® IV y el placebo en la semana 16 fue -2.8 frente al -0.7.
Se solicitó a los pacientes que indicaran el grado en que su enfermedad impactó su productividad usando una escala VAS de 0 a 10 (sin impacto en la productividad (0) a un impacto alto en la productividad (10)). En el estado basal, las puntuaciones de la mediana del impacto de la enfermedad en la productividad diaria (VAS) en el grupo tratado con SIMPONI® IV y en el grupo tratado con el placebo fue 7.6 y 7.3. La disminución de la mediana (mejoría) desde el estado basal en la puntuación del impacto de la enfermedad en la productividad diaria (VAS) en los grupos con SIMPONI® IV y el placebo en la semana 16 fue de -2.6 frente al -0.7.
Mejoría del sueño:
Estudio de EA: En el estado basal, los pacientes en el estudio en EA tuvieron alteración del sueño según lo medido con el Cuestionario de Jenkins para la Evaluacion del Sueño de 20 puntos. Los pacientes en el grupo de SIMPONI® 50 mg y en el grupo del placebo tuvieron una mediana similar de la puntuacion en el estado basal de 10.0 y 9.0, respesctivamente, indicando niveles similares de la alteración del sueño. Los pacientes tratados con SIMPONI® 50 mg mostraron una mediana de la mejoria significativamente mayor desde el estado basal de la puntuación del sueño comparado con el placebo en la semana 14 (-3.0 frente a 0.0; p<0.001) y en la semana 24 (-3.0 frente a -1.0; p<0.001). La media de la mejoría del sueño fue mayor en los pacientes tratados con SIMPONI® 50 mg en comparación con el grupo del placebo en la semana 14 (-3.1 ± 4.4 frente a -0.5 ± 3.8). Una puntuación negativa/decreciente es indicativo de la mejoría. La media de la mejoria del sueño fue sostenida por los pacientes tratados con SIMPONI® 50 mg hasta la semana 24 (Tabla 57).
|
Tabla 57: Estudio en EA: Cuestionario de Jenkins para la Evaluacion del Sueño en pacientes aleatorizados |
||
|
Placebo (N = 78)a |
SIMPONI® 50 mg* (N = 138)a |
|
|
Estado basal |
||
|
Promedio ± S.D. |
9.9 ± 4.7 |
10.3 ± 4.4 |
|
Mediana |
9.0 |
10.0 |
|
Mejoría en la semana 14 |
||
|
Promedio ± S.D. |
-0.5 ± 3.8 |
-3.1 ± 4.4 |
|
Mediana |
0.0 |
-3.0 |
|
Mejoría en la semana 24 |
||
|
Promedio ± S.D. |
-0.6 ± 4.0 |
-3.3 ± 4.2 |
|
Mediana |
-1.0 |
-3.0 |
|
*p<0.001 para todas la comparaciones. a N refleja los pacientes aleatorizados; sin embargo, N puede variar en los diferentes puntos cronológicos. El Cuestionario de Jenkins para la Evaluacion del Sueño evalúa el problema del sueño. Los pacientes indican el número de días que habian experimentado cada uno de los 4 problemas durante los últimos 30 días. Las evaluaciones luego categorizan el número de días en una escala de 0 a 5, de manera que la puntuación es de 0 (mejor) a 20 (peor). Una puntuación negativa/decreciente es indicativo de mejoria. |
||
Estudio por vía IV de EA: En el estudio por vía IV de EA, el grado de problemas para dormir se evaluó utilizando la Escala del sueño del Estudio de resultados médicos (MOS-SS), la cual midió 6 aspectos del sueño, incluyendo trastornos del sueño, somnolencia, adecuación del sueño, ronquidos, falta de aire o dolor de cabeza y las horas de sueño/sueño óptimo. En el estado basal, la mediana de la puntuación del índice de problemas de sueño fue 39 en los grupos con SIMPONI® IV y el placebo. En la semana 16, la mejoría de la mediana en el índice de problemas de sueño fue mayor para los pacientes tratados con SIMPONI® IV en comparación con el grupo del placebo (7.0 frente al 2.8). Un incremento desde el estado basal representa mejoría (Tabla 58).
|
Tabla 58: Estudio por vía IV de EA: Puntuación del índice de problemas de sueño (MOS-SS); pacientes aleatorizados |
||
|
Placebo (N=103)a |
SIMPONI® IV (N=105)a |
|
|
Puntuación compuesta en el estado basal |
||
|
Media ± DE |
39 (8.3) |
40 (7.8) |
|
Mediana |
39 |
39 |
|
Mejoría en la semana 16 |
||
|
Media ± DE |
2.5 (8.16) |
6.6 (7.18) |
|
Mediana |
2.8 |
7.0 |
|
a N refleja a los pacientes aleatorizados; sin embargo, N puede variar en diferentes puntos de evaluación. La Escala del sueño del estudio de resultados médicos (MOS-SS) mide 6 aspectos del sueño, incluyendo tratornos de sueño, somnolencia, adecuación del sueño, ronquidos, falta de aire o dolor de cabeza y horas de sueño/sueño óptimo. |
||
Espondiloartritis axial no radiográfica: La seguridad y eficacia de SIMPONI® 50 mg fueron evaluadas en un estudio (GO AHEAD) multicéntrico, aleatorizado, doble ciego, controlado con placebo (hasta la semana 16) evaluando el tratamiento con SIMPONI® 50 mg administrado por vía subcutánea cada 4 semanas en 197 pacientes adultos con nr Axial SpA activa severa (definida como aquellos pacientes que cumpliendo los criterios de clasificación ASAS para la espondiloartritis axial no cumplian con los criterios de Nueva York modificado para la EA). En la semana 16, los pacientes ingresaron en un período abierto en el que todos los pacientes recibieron SIMPONI® 50 mg administrado por vía subcutánea cada 4 semanas hasta la semana 48 con evaluaciones de la eficacia realizadas hasta la semana 52 y del seguimiento de la seguridad hasta la semana 60. Aproximadamente el 93% de los pacientes que estaban recibiendo SIMPONI® al inicio de la extensión del periodo abierto (semana 16) permanecieron en el tratamiento hasta el final del estudio (semana 52).
Los pacientes incluidos en este estudio tenían enfermedad activa, definida como un índice BASDAI ≥ 4 cm y una escala VAS para el dolor de espalda total ≥ 4 cm, cada uno en una escala de 0-10 cm, y experimentaron una respuesta inadecuada al tratamiento con AINEs o fueron intolerantes al tratamiento con AINEs.
Los pacientes que tenían sacroileítis bilateral de Grado 2 o sacroileítis unilateral de Grado 3 o Grado 4 en las radiografías convencionales fueron excluidos del estudio. Los pacientes que previamente habían recibido bloqueadores-TNFa o o cualquier agente biológico también fueron excluidos del estudio.
Los pacientes fueron aleatorizados para recibir placebo (n=100) o SIMPONI® 50 mg (n=97). El criterio de evaluación principal fue la respuesta ASAS 20 en la semana 16. Los principales criterios de evaluación secundaria incluyeron respuesta ASAS 40 en la semana 16, respuesta BASDAI 50 en la semana 16, remisión parcial ASAS en la semana 16, y el cambio de la puntuación del Consorcio de Investigación de la Espondiloartritis de Canadá (SPARCC) determinada mediante las MRI de las articulaciones sacroiliacas desde el estado basal hasta la semana 16.
Los análisis se realizaron en toda la población tratada (AT N=197) y en la población con Signos Objetivos de Inflamación (OSI, N=158). La población con OSI fue definida por una elevada PCR por encima del límite superior de la evidencia normal y/o por la evidencia real de sacroileítis en las MRI en el estado basal.
Reducción de signos y síntomas: El tratamiento con SIMPONI® 50 mg causó una mejoría de los signos y síntomas según lo demostrado por los principales criterios de evaluación en la semana 16 (Tabla 59). Una proporción significativamente mayor de pacientes alcanzaron ASAS 20, ASAS 40, ASAS 5/6 e índice BASDAI 50 en el grupo de SIMPONI® 50 mg en comparación con el grupo del placebo (p<0.0001). Además, una proporción significativamente mayor de pacientes alcanzaron remisión parcial ASAS y ASDAS-C <1.3 en el grupo de SIMPONI® 50 mg en comparación con el placebo (p<0.05). Se observó una mejoria significativa en ASAS 20 ya en la primera evaluación (semana 4, p<0.0001) después de la administración inicial de SIMPONI® 50 mg (Figura 10).
Figura 10: Porcentaje de sujetos que alcanzaron respuesta ASAS 20 por tiempo cronológico en toda la población tratada (AT)
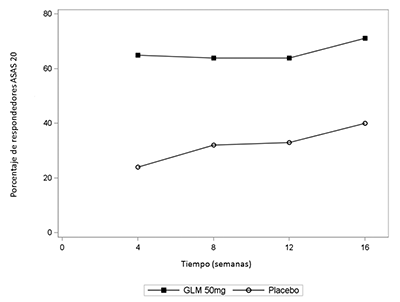
Resultados significativos para estos mismos criterios de evaluación también fueron demostrados en la población con OSI (Tabla 59). Se observaron mejoras similares en todos los principales criterios de evaluación descritos anteriormente desde la semana 16 hasta la semana 52 entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI® 50 mg en toda la problación tratada y en la población con OSI.
|
Tabla 59: Estudio de nr Axial SpA: Porcentaje de pacientes con nr Axial SpA con respuesta ASAS, BASDAI y ASDAS-C en pacientes aleatorizados y tratados |
||
|
AT |
Placebo |
SIMPONI® 50mg |
|
ASAS 20 (% de respondedores) |
||
|
Semana 16 |
40% |
71%** |
|
ASAS 40 (% de respondedores) |
||
|
Semana 16 |
23% |
57%** |
|
Remisión parcial ASAS (% de respondedores) |
||
|
Semana 16 |
18% |
33%* |
|
ASAS 5/6a (% de respondedores) |
||
|
Semana 16 |
23% |
54%** |
|
BASDAI 50 (% de respondedores) |
||
|
Semana 16 |
30% |
58%** |
|
ASDAS-Cab < 1.3 (% de respondedores) |
||
|
Semana 16 |
13% |
33%* |
|
OSI |
Placebo |
SIMPONI® 50mg* |
|
ASAS 20 (% de respondedores) |
||
|
Semana 16 |
38% |
77% ** |
|
ASAS 40 (% de respondedores) |
||
|
Semana 16 |
23% |
60%** |
|
Remisión parcial ASAS (% de respondedores) |
||
|
Semana 16 |
19% |
35%* |
|
ASAS 5/6a (% de respondedores) |
||
|
Semana 16 |
23% |
63%** |
|
BASDAI 50 (% de respondedores) |
||
|
Semana 16 |
29% |
59%** |
|
ASDAS-Cab < 1.3 (% de respondedores) |
||
|
Semana 16 |
16% |
35%* |
|
* p <0.05 para comparaciones de SIMPONI® 50 mg frente al placebo ** p<0.0001para comparaciones de SIMPONI® 50 mg frente al placebo a No controlado para error del Tipo I b Puntuación de la Actividad de la Enfermedad Espondilitis Anquilosante conteniendo Proteína C-Reactiva (AT-Placebo, N=90; AT- SIMPONI® 50 mg, N=88; OSI-Placebo, N=71; OSI- SIMPONI® 50 mg, N=71) Una respuesta ASAS 20 (Anderson et al, 2001) fue definida como: 1. Una mejoría ≥ 20% desde el estado basal y una mejoría absoluta desde el estado basal de por lo menos 1 en una escala de 0 a 10 cm en por lo menos 3 de los siguientes 4 parámetros: Evaluación general del paciente, Evaluación del dolor (dolor de espalda total), Puntuación de índice BASFI, o Inflamación (promedio de las 2 preguntas del índice BASDAI relativas a la rigidez matutina). 2. Ausencia de deterioro desde el estado basal (deterioro definido como ≥ 20% del empeoramiento y empeoramiento absoluto de por lo menos 1 en una escala de 0 a 10 cm) en el parámetro potencial restante. ASAS 40: Un ASAS 40 se define como ≥ 40% de mejoría en 3 de los 4 parámetros, con una mejoría absoluta de por lo menos 2 en una escala de 0 a 10 cm, y sin empeoramiento en el parámetro restante. Remisión parcial ASAS: Cada uno de los 4 parámetros del ASAS 20 tiene una puntuación inferior a 2. ASAS 5/6 se define como el 20% o más de mejoria en uno de los 5 de los 6 parámetros del dolor de espalda total en la escala VAS, paciente en general en la escala VAS, función (BASFI), rigidez matutina (promedio de las últimas 2 preguntas del índice BASDAI), proteína C-reactiva y la medida de la flexión lateral lumbar BASDAI: Autoevaluación del sujeto utilizando una escala VAS (0 a 10 cm) con los siguientes criterios: Fatiga, dolor de columna, dolor en las articulaciones, entesitis, rigidez matutina cualitativa y rigidez matutina cuantitativa. |
||
Los pacientes tratados con SIMPONI® 50 mg alcanzaron mejoria significativamente mayor en comparación con el placebo en la población AT y la población con OSI en la evaluación general del paciente, dolor de espalda total, inflamación, dolor de espalda nocturna, proteína C-reactiva y ASDAS-PCR (Tabla 60). Se observaron mejorias similares en estos criterios de evaluación hasta la semana 52 entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI® 50 mg en la población AT y la población con OSI.
|
Tabla 60: Estudio de nr Axial SpA: Mejoría de los signos y sintomas en pacientes aleatorizados y tratados |
|||
|
AT |
Placebo |
SIMPONI® 50 mg |
valor-p |
|
Evaluación general del paciente de la actividad de la enfermedada (cm): cambio desde el estado basal |
|||
|
N b |
96 |
93 |
|
|
Estado basal (mediana) |
6.4 |
7.3 |
|
|
Semana 16 (mediana) |
-1.1 |
-4.2 |
|
|
Semana 16 (media ± S.E.) |
-1.5 ± 0.3 |
-3.7 ± 0.3 |
p<0.0001 |
|
Dolor de espalda totala (cm): cambio desde el estado basal |
|||
|
N b |
97 |
93 |
|
|
Estado basal (mediana) |
6.4 |
7.2 |
|
|
Semana 16 (mediana) |
-1.5 |
-4.2 |
|
|
Semana 16 (media ± S.E.) |
-2.0 ± 0.3 |
-4.1 ± 0.3 |
p<0.0001 |
|
Inflamacióna,c (cm): cambio desde el estado basal de la rigidez matutina |
|||
|
N b |
96 |
93 |
|
|
Estado basal (mediana) |
5.8 |
7.1 |
|
|
Semana 16 (mediana) |
-1.3 |
-4.0 |
|
|
Semana 16 (media ± S.E.) |
-1.8 ± 0.2 |
-3.8 ± 0.2 |
p<0.0001 |
|
Dolor de espalda nocturnaa (cm): cambio desde el estado basal |
|||
|
N b |
97 |
93 |
|
|
Estado basal (mediana) |
6.5 |
7.8 |
|
|
Semana 16 (mediana) |
-0.7 |
-5.2 |
|
|
Semana 16 (media ± S.E.) |
-1.7 ± 0.3 |
-4.4 ± 0.3 |
p<0.0001 |
|
Proteína C-reactivad (mg/dL): cambio desde el estado basal |
|||
|
N b |
91 |
88 |
|
|
Estado basal (mediana) |
0.5 |
0.4 |
|
|
Semana 16 (mediana) |
0.0 |
-0.0 |
|
|
Semana 16 (media ± S.E.) |
-0.4 ± 0.2 |
-1.0 ± 0.2 |
p=0.0003 |
|
Puntuación ASDAS-PCR: cambio desde el estado basal |
|||
|
N b |
90 |
88 |
|
|
Estado basal (mediana) |
3.3 |
3.5 |
|
|
Semana 16 (mediana) |
-0.6 |
-1.7 |
|
|
Semana 16 (media ± S.E.) |
-0.6 ± 0.1 |
-1.7 ± 0.1 |
p<0.0001 |
|
OSI |
Placebo |
SIMPONI® 50 mg |
valor-p |
|
Evaluación general del paciente de la actividad de la enfermedada (cm): cambio desde el estado basal |
|||
|
N b |
77 |
76 |
|
|
Estasdo basal (mediana) |
6.6 |
7.4 |
|
|
Semana 16 (mediana) |
-0.8 |
-4.4 |
|
|
Semana 16 (media ± S.E.) |
-1.4 ± 0.4 |
-3.9 ± 0.4 |
p<0.0001 |
|
Dolor de espalda totala (cm): cambio desde el estado basal |
|||
|
N b |
78 |
76 |
|
|
Estado basal (mediana) |
6.5 |
7.3 |
|
|
Semana 16 (mediana) |
-1.3 |
-4.4 |
|
|
Semana 16 (media ± S.E.) |
-1.9 ± 0.3 |
-4.2 ± 0.3 |
p<0.0001 |
|
Inflamacióna,c (cm): cambio desde el estado basal en la rigidez matutina |
|||
|
N b |
77 |
76 |
|
|
Estado basal (mediana) |
5.9 |
6.9 |
|
|
Semana 16 (mediana) |
-1.3 |
-4.0 |
|
|
Semana 16 (media ± S.E.) |
-1.7 ± 0.3 |
-4.0 ± 0.3 |
p<0.0001 |
|
Dolor de espalda nocturnoa (cm): cambio desde el estado basal |
|||
|
N b |
78 |
76 |
|
|
Estado basal (mediana) |
6.8 |
7.9 |
|
|
Semana 16 (mediana) |
-0.8 |
-5.6 |
|
|
Semana 16 (media ± S.E.) |
-1.8 ± 0.3 |
-4.7 ± 0.3 |
p<0.0001 |
|
Proteína C-reactivad (mg/dL): cambio desde el estado basal |
|||
|
N b |
72 |
71 |
|
|
Estado basal (mediana) |
0.8 |
0.7 |
|
|
Semana 16 (mediana) |
0.0 |
-0.4 |
|
|
Semana 16 (media ± S.E.) |
-0.50 ± 0.2 |
-1.2 ± 0.2 |
p=0.0004 |
|
Puntuación ASDAS-CRP: cambio desde el estado basal |
|||
|
N b |
71 |
71 |
|
|
Estado basal (mediana) |
3.4 |
3.6 |
|
|
Semana 16 (mediana) |
-0.5 |
-2.0 |
|
|
Semana 16 (media ± S.E.) |
-0.6 ± 0.1 |
-1.8 ± 0.1 |
p<0.0001 |
|
a Escala Analógica Visual (con 0 = “mejor” y 10 = “peor”). Una puntuación negativa/decreciente es indicativo de mejoría b N refleja los pacientes con datos en el estado basal y en la semana 16 c Promedio de las últimas 2 preguntas de las 6 preguntas del índice BASDAI d Rango normal de 0-0.9 mg/dL ASDAS-PCR: Puntuación del Índice de la Actividad de la Enfermedad Espondilitis Anquilosante conteniendo PCR |
|||
La entesitis se evaluó mediante la puntuación del índice Maastricht para la Entesitis en Espondilitis Anquilosante (MASES). Se demostró mejoría media estadísticamente significativa desde el estado basal de la puntuación de la entesitis medida por el índice MASES en la semana 16 en la población AT (Tabla 61). Se observaron respuestas similares del índice MASES en la semana 24 y en la semana 52 entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI® 50 mg en la población AT y la problación con OSI.
|
Tabla 61: Estudio de nr Axial SpA: Puntuación de la Entesitis en pacientes aleatorizados y tratados |
|||
|
AT |
Placebo |
SIMPONI® 50 mg |
valor-p |
|
Índice MASES (rango 0-13): cambio desde el estado basal |
|||
|
N a |
97 |
92 |
|
|
Estado basal (mediana) |
2.0 |
2.0 |
|
|
Semana 16 (mediana) |
0.0 |
-1.0 |
|
|
Semana 16 (media ± S.E.) |
-0.7 ± 0.3 |
-1.4 ± 0.3 |
p=0.0302 |
|
OSI |
Placebo |
SIMPONI® 50 mg |
valor-p |
|
Índice MASES (rango 0-13): cambio desde el estado basal |
|||
|
N a |
77 |
75 |
|
|
Estado basal (mediana) |
2.0 |
2.0 |
|
|
Semana 16 (mediana) |
0.0 |
-1.0 |
|
|
Semana 16 (media ± S.E.) |
-0.8 ± 0.3 |
-1.4 ± 0.3 |
NS |
|
a N refleja a los pacientes con datos en el estado basal y en la semana 16. MASES: Índice Maastricht para la Entesitis en Espondilitis Anquilosante: Existen 13 sitios que estan siendo medidos. Todos los sitios tiene como puntuación 0 o 1 (0 no doloroso, 1 doloroso). El índice MASES es la suma de las puntuaciones de todos los sitios (de 0 a 13). |
|||
Mejoría en el rango de movimiento: El rango de movimiento y la movilidad de la columna fueron evaluados por BASMI (Índice de metrología de Bath para Espondilitis Anquilosante) y la expansión de la pared torácica en el estado basal y en la semana 16. Se observó una media de la mejoría estadísticamente significativa en BASMI en la semana 16 en el grupo de SIMPONI® 50 mg en comparación con el grupo del placebo. También se observó una mejoría estadísticamente significativa en BASMI en la población con OSI en el grupo de SIMPONI® 50 mg en comparación con el grupo del placebo (Tabla 62). Se observaron mejorías similares de BASMI en la semana 24 y en la semana 52 entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI® 50 mg en las población AT y la población con OSI.
|
Tabla 62: Estudio de nr Axial SpA: Análisis de la movilidad de la columna en pacientes aleatorizados y tratados |
|||
|
AT |
Placebo |
SIMPONI® 50 mg |
valor-p |
|
BASMI (0-10 cm): cambio desde el estado basal |
|||
|
N a |
100 |
94 |
|
|
Estado basal (mediana) |
2.2 |
2.2 |
|
|
Semana 16 (mediana) |
0.0 |
-0.4 |
|
|
Semana 16 (media ± S.E.) |
-0.1 ± 0.1 |
-0.5 ± 0.1 |
p<0.0001 |
|
Expansión de la pared torácica (cm): cambio desde el estado basal |
|||
|
N a |
97 |
90 |
|
|
Estado (mediana) |
5.0 |
5.0 |
|
|
Semana 16 (mediana) |
0.0 |
0.5 |
|
|
Semana 16 (media ± S.E.) |
0.0 ± 0.2 |
0.3 ± 0.2 |
NS |
|
OSI |
Placebo |
SIMPONI® 50 mg |
valor-p |
|
BASMI (0-10 cm): cambio desde el estado basal |
|||
|
N a |
80 |
77 |
|
|
Estado basal (mediana) |
2.0 |
2.2 |
|
|
Semana 16 (mediana) |
0.0 |
-0.4 |
|
|
Semana 16 (media ± S.E.) |
-0.1 ± 0.1 |
-0.5 ± 0.1 |
p=0.0002 |
|
Expansión de la pared torácica (cm): cambio desde el estado basal |
|||
|
N a |
78 |
73 |
|
|
Estado basal (mediana) |
4.6 |
5.0 |
|
|
Semana 16 (mediana) |
0.0 |
0.3 |
|
|
Semana 16 (media ± S.E.) |
0.0 ± 0.2 |
0.3 ± 0.2 |
NS |
|
a N refleja a los pacientes con datos en el estado basal y la semana 16. Valores normales de la movilidad de la columna: expansión torácica: > 6.5 cm en varones, > 4.5 cm en mujeres, de 35-45 años. |
|||
Inhibición de la inflamación en las MRI: El análisis de la puntuación de las articulaciones sacroilíacas en las MRI por el SPARCC (Consorcio de Investigación de la Espondiloartritis en Canadá) desde el estado basal hasta la semana 16 se realizó en sujetos con mediciones de las MRI en el estado basal y en la semana 16. En el grupo de sujetos que completaron la semana 16, se observó una disminución significativamente mayor desde el estado basal en los sujetos que recibieron SIMPONI® 50 mg en comparación con los que recibieron el placebo (p<0.0001). También se observaron resultados estadísticamente significativos en la población con OSI (Tabla 63).
|
Tabla 63: Inhibición de la inflamación en las articulaciones sacroilíacas, medida por el cambio medio desde el estado basal en las MRI |
||
|
AT |
Placebo (N=87)a |
SIMPONI® 50 mg (N=74)a |
|
Puntuación de la articulación sacroilíaca mediante MRI por el SPARCC b |
-0.9 |
-5.3* c |
|
OSI |
Placebo (N=69)a |
SIMPONI® 50 mg (N=61)a |
|
Puntuación de la articulación sacroilíaca mediante MRI por el SPARCC |
-1.2 |
-6.4* c |
|
*p <0.0001 para comparaciones de SIMPONI® 50 mg frente al placebo a N refleja los pacientes con datos en el estado basal y en la semana 16 b Consorcio de Investigación de la Espondiloartritis de Canadá c Basado en la prueba de Mann-Whitney |
||
Mejoría de la función física: La función física fue evaluada por el índice BASFI (Índice de funcionalidad Bath de la Espondilitis Anquilosante) en el estado basal y en la semana 16. Los pacientes tratados con SIMPONI® 50 mg reportaron mejorías estadísticamente significativas de la función física en comparación con el placebo. En la población con OSI, también se observó una media de la mejoría en el índice BASFI en la semana 16 en el grupo de SIMPONI® 50 mg en comparación con el grupo del placebo (Tabla 64). Se observaron mejoras similares del índice BASFI hasta la semana 52 entre los pacientes que permanecieron en el estudio y en tratamiento con SIMPONI® 50 mg en la población AT y la población con OSI.
|
Tabla 64: Estudio en nr Axial SpA: Mejoría en el análisis del índice BASFI en pacientes aleatorizados y tratados |
|||
|
AT |
Placebo (N=97)a |
SIMPONI® 50 mg (N=93)a |
valor-p |
|
BASFI (0-10 cm): cambio desde el estado basal |
|||
|
Estado basal (mediana) |
5.0 |
5.2 |
|
|
Semana 16 (mediana) |
-0.4 |
-2.6 |
|
|
Semana 16 (media ± S.E.) |
-0.9 ± 0.2 |
-2.6 ± 0.2 |
p<0.0001 |
|
OSI |
Placebo (N=78)a |
SIMPONI® 50 mg (N=76)a |
valor-p |
|
BASFI (0-10 cm): cambio desde el estado basal |
|||
|
Estado basal (mediana) |
5.1 |
5.2 |
|
|
Semana 16 (mediana) |
-0.4 |
-2.9 |
|
|
Semana 16 (media ± S.E.) |
-0.9 ± 0.2 |
-2.8 ± 0.3 |
p<0.0001 |
|
a N refleja los pacientes con datos en el estado basal y en la semana 16. Índice de funcionalidad Bath de la Espondilitis Anquilosante (BASFI), promedio de 10 preguntas. Una puntuación negativa/decreciente es indicativo de mejoría en el índice BASFI. |
|||
Mejoría en la calidad de vida relacionada con la salud: La calidad de vida relacionada con la salud fue medida por el SF-36 (Cuestionario de Salud de formato corto 36), los Resúmenes de los Componentes Físicos y Mentales, el AsQoL (Cuestionario de Calidad de Vida de Espondilitis Anquilosante), la Puntuación del Índice EQ-5D (Cuestionario EuroQoL de 5 Dimensiones), la Puntuación del Estado de Salud del EQ-5D y la WPAI (Encuesta sobre Productividad Laboral y Deterioro de las Actividades) en el estado basal, semana 16 (Tabla 65) y semana 52.
Los pacientes tratados con SIMPONI® 50 mg mostraron una media de la mejoría estadísticamente significativa desde el estado basal de la puntuación del resumen del componente físico (PCS) del SF-36, en comparación con el placebo en la semana 16 (Tabla 65). La mejora observada en la semana 16 entre los pacientes tratados con SIMPONI® 50 mg continuó en los pacientes que permanecieron en el estudio en la semana 52.
En la puntuación del resumen del componente mental (MCS) del SF-36, los pacientes tratados con SIMPONI® 50 mg mostraron una media de la mejoría estadísticamente significativa desde el estado basal, en comparación con el placebo, en la semana 16 (Tabla 65). La mejora observada en la semana 16 entre los pacientes tratados con SIMPONI® 50 mg continuó en los pacientes que permanecieron en el estudio en la semana 52.
En la puntuación del ASQoL, los pacientes tratados con SIMPONI® 50 mg mostraron una media de la mejoría significativa mayor desde el estado basal, en comparación con el placebo en la semana 16 (Tabla 65). La mejoría observada en la semana 16 entre los pacientes tratados con SIMPONI® 50 mg continuó en los pacientes que permanecieron en el estudio en la semana 52.
En la puntuación del Índice EQ-5D, los pacientes tratados con SIMPONI® 50 mg mostraron una media de la mejoría significativamente mayor desde el estado basal, en comparación con el placebo en la semana 16. La mejoría observada en la semana 16 entre los pacientes tratados con SIMPONI® 50 mg continuó en los pacientes que permanecieron en el estudio en la semana 52.
En la puntuación del Estado de Salud del EQ-5D, los pacientes tratados con SIMPONI® 50 mg mostraron una media de la mejoría estadísticamente mayor significativa desde el estado basal, en comparación con el placebo en la semana 16 (Tabla 65). La mejoría observada en la semana 16 entre los pacientes tratados con SIMPONI® 50 mg continuó en los pacientes que permanecieron en el estudio en la semana 52.
También se observó una media de la mejoría estadísticamente significativa en la población con OSI en todas las mediciones de calidad de vidad relacionada con la salud descritas anteriormente (Tabla 65) en la semana 16. La mejoría observada en la semana 16 entre los pacientes tratados con SIMPONI® 50 mg en la población con OSI continuó en los pacientes que permanecieron en el estudio en la semana 52.
La productividad en el trabajo y dentro del hogar fueron evaluadas en el estado basal y en la semana 16 según lo reportado por las encuestas WPAI que miden el porcentaje de tiempo laboral perdido debido a la salud, el porcentaje de discapacidad mientras se trabaja debido a la salud, el porcentaje de discapacidad general debido a la salud y el porcentaje de dispacidad de la actividad debido a la Salud. Los pacientes tratados con SIMPONI® 50 mg reportaron reducciones significativamente mayores en la discapacidad general laboral y en la discapacidad de la actividad, en comparación con el placebo. En la población con OSI, también se observaron reducciones estadísticamente significativas en la discapacidad durante el trabajo, discapacidad general laboral y en la discapacidad de la actividad (Tabla 65).
Las reducciones observadas en la semana 16 entre los pacientes tratados con SIMPONI® 50 mg en la población AT y la población con OSI y continuaron en los pacientes que permanecieron en el estudio en la semana 52.
|
Tabla 65: Estudio de nr Axial SpA: Análisis de la calidad de vidad relacionada con la salud en pacientes aleatorizados y tratados |
|||
|
AT |
Placebo |
SIMPONI® 50 mg |
valor-p |
|
PCS del SF-36 |
|||
|
N a |
96 |
91 |
|
|
Estado basal |
|||
|
Media ± S.D. |
35.0 ± 8.7 |
32.9 ± 8.1 |
|
|
Mediana |
35.6 |
33.0 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.E. b |
3.7 ± 0.8 |
10.3 ± 0.8 |
p<0.0001 |
|
Mediana |
2.5 |
9.7 |
|
|
MCS del SF-36 |
|||
|
N a |
96 |
91 |
|
|
Estado basal |
|||
|
Media ± S.D. |
41.6 ± 11.1 |
41.1 ± 11.9 |
|
|
Mediana |
41.4 |
41.7 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.E. b |
1.6 ± 1.1 |
5.9 ± 1.1 |
p=0.0034 |
|
Mediana |
2.0 |
4.9 |
|
|
ASQoL |
|||
|
N a |
100 |
94 |
|
|
Estado basal |
|||
|
Media ± S.D. |
10.2 ± 4.6 |
11.1 ± 4.5 |
|
|
Mediana |
11.0 |
12.0 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.E. b |
-1.8 ± 0.5 |
-5.2 ± 0.5 |
p<0.0001 |
|
Mediana |
-1.0 |
-4.0 |
|
|
Puntuación de Índice EQ-5D |
|||
|
N a |
100 |
94 |
|
|
Estado basal |
|||
|
Media ± S.D. |
0.4 ± 0.3 |
0.4 ± 0.3 |
|
|
Mediana |
0.5 |
0.6 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.E. b |
0.1 ± 0.0 |
0.3 ± 0.0 |
p<0.0001 |
|
Mediana |
0.1 |
0.2 |
|
|
Puntuación del estado de salud del EQ-5D en la escala VAS (cm) |
|||
|
N a |
100 |
94 |
|
|
Estado basal |
|||
|
Media ± S.D. |
5.1 ± 2.1 |
4.5 ± 2.2 |
|
|
Mediana |
5.0 |
4.5 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.E. b |
0.6 ± 0.2 |
2.1 ± 0.2 |
p<0.0001 |
|
Mediana |
0.0 |
2.0 |
|
|
WPAI: Porcentaje del tiempo de trabajo perdido debido a la Salud |
|||
|
N a |
58 |
58 |
|
|
Estado basal |
|||
|
Media ± S.D. |
13.4 ± 25.8 |
11.3 ± 24.0 |
|
|
Mediana |
0 |
0 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.D. b |
0.9 ± 27.9 |
-5.8 ± 16.3 |
NS |
|
Mediana |
0 |
0 |
|
|
WPAI: Porcentaje de discapacidad mientras se trabaja debido a la salud |
|||
|
N a |
58 |
63 |
|
|
Estado basal |
|||
|
Media ± S.D. |
49.3 ± 26.4 |
43.7 ± 29.0 |
|
|
Mediana |
50.0 |
40.0 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.D. b |
-14.0 ± 28.7 |
-22.4 ± 25.8 |
NS |
|
Mediana |
-10.0 |
-20.0 |
|
|
WPAI: Porcentaje de discapacidad laboral general debido a la Salud |
|||
|
N a |
57 |
57 |
|
|
Estado basal |
|||
|
Media ± S.D. |
53.2 ± 27.1 |
43.0 ± 29.9 |
|
|
Mediana |
53.9 |
40.0 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.D. b |
-11.7 ± 28.2 |
-21.1 ± 24.7 |
p=0.0391 c |
|
Mediana |
-8.0 |
-20.0 |
|
|
WPAI: Porcentaje de discapacidad de la actividad debido a la Salud |
|||
|
N a |
100 |
93 |
|
|
Estado basal |
|||
|
Media ± S.D. |
54.4 ± 24.7 |
55.1 ± 25.7 |
|
|
Mediana |
60.0 |
60.0 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.D. b |
-8.6 ± 27.6 |
-24.9 ± 29.5 |
p<0.0001 c |
|
Mediana |
-10.0 |
-20.0 |
|
|
OSI |
Placebo |
SIMPONI® 50 mg |
valor-p |
|
PCS del SF-36 |
|||
|
N a |
78 |
74 |
|
|
Estado basal |
|||
|
Media ± S.D. |
35.1 ± 8.9 |
33.0 ± 8.3 |
|
|
Mediana |
35.1 |
33.1 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.E. b |
3.7 ± 0.9 |
10.4 ± 0.9 |
p<0.0001 |
|
Mediana |
2.5 |
9.7 |
|
|
MCS del SF-36 |
|||
|
N a |
78 |
74 |
|
|
Estado basal |
|||
|
Media ± S.D. |
42.2 ± 11.0 |
40.9 ± 11.4 |
|
|
Mediana |
42.1 |
41.1 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.E. b |
1.0 ± 1.2 |
5.6 ± 1.3 |
p=0.0065 |
|
Mediana |
0.7 |
4.9 |
|
|
ASQoL |
|||
|
N a |
80 |
77 |
|
|
Estado basal |
|||
|
Media ± S.D. |
10.2 ± 4.8 |
10.9 ± 4.4 |
|
|
Mediana |
11.0 |
12.0 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.E. b |
-1.8 ± 0.5 |
-5.1 ± 0.5 |
p<0.0001 |
|
Mediana |
-1.0 |
-5.0 |
|
|
Puntuación de Índice EQ-5D |
|||
|
N a |
80 |
77 |
|
|
Estado basal |
|||
|
Media ± S.D. |
0.4 ± 0.3 |
0.4 ± 0.3 |
|
|
Mediana |
0.5 |
0.6 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.E. b |
0.1 ± 0.0 |
0.3 ± 0.0 |
p=0.0004 |
|
Mediana |
0.1 |
0.2 |
|
|
Puntuación del estado de salud del EQ-5D en la escala VAS (cm) |
|||
|
N a |
80 |
77 |
|
|
Estado basal |
|||
|
Media ± S.D. |
5.2 ± 2.1 |
4.6 ± 2.2 |
|
|
Mediana |
5.0 |
4.7 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.E. b |
0.5 ± 0.3 |
2.0 ± 0.3 |
p<0.0001 |
|
Mediana |
0.0 |
2.0 |
|
|
WPAI: Porcentaje del tiempo de trabajo perdido debido a la salud |
|||
|
N a |
46 |
47 |
|
|
Estado basal |
|||
|
Media ± S.D. |
15.5 ± 28.2 |
12.8 ± 26.1 |
|
|
Mediana |
0 |
0 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.D. b |
2.1 ± 30.9 |
-6.0 ± 17.6 |
NS |
|
Mediana |
0 |
0 |
|
|
WPAI: Porcentaje de discapacidad mientras se trabaja debido a la salud |
|||
|
N a |
46 |
51 |
|
|
Estado basal |
|||
|
Media ± S.D. |
50.4 ± 26.8 |
45.9 ± 28.4 |
|
|
Mediana |
50.0 |
50.0 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.D. b |
-11.7 ± 29.8 |
-23.3 ± 26.5 |
p=0.0194 c |
|
Mediana |
-10.0 |
-20.0 |
|
|
WPAI: Porcentaje de discapacidad laboral general debido a la Salud |
|||
|
N a |
45 |
46 |
|
|
Estado basal |
|||
|
Media ± S.D. |
54.6 ± 27.6 |
45.5 ± 29.3 |
|
|
Mediana |
58.3 |
45.0 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.D. b |
-8.4 ± 28.5 |
-21.2 ± 24.7 |
p=0.0090 c |
|
Mediana |
-4.8 |
-20.0 |
|
|
WPAI: Porcentaje de discapacidad de la actividad debido a la salud |
|||
|
N a |
80 |
76 |
|
|
Estado basal |
|||
|
Media ± S.D. |
52.9 ± 24.6 |
57.5 ± 24.3 |
|
|
Mediana |
55.0 |
60.0 |
|
|
Semana 16, mejoría desde el estado basal |
|||
|
Media ± S.D. b |
-6.8 ± 28.9 |
-25.5 ± 28.7 |
p<0.0001 c |
|
Mediana |
-10.0 |
-20.0 |
|
|
a N indica los pacientes con datos en el estado basal y en la semana 16. b Derivado de modelo estadístico c Basado en la prueba Mann-Whitney El valor promedio de la puntuación del componente físico (PCS) y la puntuación del componente mental (MCS) del SF-36 es 50 ± 10 cada uno para la población general de Estados Unidos. El rango es de 0 (peor) a 100 (mejor). ASQoL: El Cuestionario de Calidad de Vida de Espondilitis Anquilosante es un instrumento específico de la enfermedad que se utiliza para medir la calidad de vida en la población de pacientes con EA. Consta de 18 preguntas de sí o no como respuesta a las preguntas relacionadas con el impacto del dolor sobre el sueño, estado de ánimo, motivación, capacidad para tolerar, actividades de la vida diaria, independencia, relaciones y vida social. EQ 5D: Cuestionario de salud EuroQol-5D que comprende 5 dimensiones: movilidad, cuidado personal, actividades habituales, dolor/malestar y ansiedad/depresión Índice EQ-5D: Puntaje ponderado (5D + VAS): WPAI: Índice de la productividad laboral y el deterioro: mide el tiempo de trabajo perdido, incapacidad para el trabajo y el deterioro de las actividades regulares debido a la salud y los síntomas en general. El WPAI se evalúa en relación con las medidas de las percepciones generales de la salud, del rol (física), del rol (emocional), el dolor, la severidad de los síntomas, medidas generales del trabajo e interferencia con la actividad regular. |
|||
Colitis ulcerativa: La seguridad y eficacia de SIMPONI® fue evaluada en dos estudios clínicos aleatorizados, multicéntricos, doble ciego, controlado con placebo en pacientes adultos ≥ 18 años de edad.
En el estudio de inducción (Estudio 1 en CU) se evaluaron pacientes con colitis ulcerativa de moderadamente a severamente activa (puntuación Mayo 6 a 12; Subpuntuación endoscópica ≥ 2) que tenían una respuesta inadecuada a las terapias convencionales o no habían logrado tolerarlas, o eran dependientes de los corticosteroides. El estudio fue un estudio combinado de Fase 2 (determinación de la dosis) y Fase 3 (confirmación de la dosis). En el periodo de determinación de la dosis del estudio, los pacientes fueron aleatorizados para formar parte de uno de los 4 grupos de tratamiento: SIMPONI® 400 mg administrado por vía SC en la semana 0 y 200 mg en la semana 2 (400/200 mg), SIMPONI® 200 mg administrado por vía SC en la semana 0 y 100 mg en la semana 2 (200/100 mg), SIMPONI® 100 mg administrado por vía SC en la semana 0 y 50 mg en la semana 2 (100/50 mg) o placebo por vía SC en la semanas 0 y en la semana 2. En el periodo de confirmación de la dosis del estudio, la eficacia fue evaluada en 761 pacientes quienes fueron aleatorizados para recibir SIMPONI® 400 mg por vía SC en la semana y 200 mg en la semana 2, SIMPONI® 200 mg por vía SC en la semana 0 y 100 mg en la semana 2 o placebo por vía SC en la semana 0 y en la semana 2. Se permitió administrar dosis estables concomitantes de aminosalicilatos orales, corticosteroides y/o agentes inmunomoduladores. El principal criterio de evaluación fue la respuesta clínica en la semana 6. Los criterios principales de evaluación secundaria fueron la remisión clínica, curación de la mucosa y mejoramiento de la puntuación en el IBDQ (Cuestionario de la enfermedad intestinal inflamatoria), todas en la semana 6.
En el estudio de mantenimiento (Estudio 2 en CU) se evaluaron 456 pacientes que alcanzaron respuesta clínica con SIMPONI® desde la inducción previa con golimumab. Los pacientes fueron aleatorizadosa para recibir SIMPONI® 50 mg, SIMPONI® 100 mg o placebo administrado por vía subcutánea cada 4 semanas. Se permitió administrar dosis estables concomitantes de aminosalicilatos orales y/o agentes inmunomoduladores. Los corticosteroides debían ser reducidos al iniciar el estudio de mantenimiento. La eficacia de SIMPONI® fue evaluada hasta la semana 54 en este estudio. El principal criterio de evaluación fue el mantenimiento de la respuesta clínica hasta la semana 54. Los principales criterios de evaluación secundaria seleccionadas incluyeron la remisión clínica en la semana 30 y la semana 54 y la curación de la mucosa en la semana 30 y en la semana 54. Los pacientes que completaron el estudio de mantenimiento hasta la semana 54 continuaron el tratamiento en una extensión del estudio, con evalución de la eficacia hasta la semana 216.
En ambos estudios, la respuesta clínica y la remisión clínica fueron definidos en base a la puntuación Mayo, el cual consiste en 4 subpuntuaciones: frecuencia de las deposiciones, sangrado rectal, hallazgos en la endoscopia y evaluación general del médico. Cada subpuntuación es calificada en una escala de 0 a 3, indicando actividad normal (0) a severa (3). La puntuación Mayo es la suma de las 4 subpuntuaciones. La respuesta clínica fue definida como una disminución desde la semana 0 de inducción de la puntuación Mayo ≥ 30% y ≥ 3 puntos, acompañado por una disminución en la subpuntuación del sangrado rectal ≥ 1 o subpuntuación del sangrado rectal de 0 o 1. La remisión clínica fue definida como una puntuación Mayo ≤ 2 puntos, sin subpuntuación individual >1. La curación de la mucosa fue definida como una subpuntuación endoscópica (de la puntuación Mayo) de 0 o 1.
La calidad de vida relacionada a la salud fue evaluada usando el IBDQ, el SF-36 y el índice EQ-5D. El IBDQ es un cuestionario específicamente diseñado para los pacientes con enfermedad inflamatoria intestinal. El SF-36 es un cuestionario del estado general de la salud que ha sido ampliamente usado en diversas enfermedades y condiciones para evaluar el bienestar mental y físico de los pacientes. El índice EQ-5D es un instrumento no específico de la enfermedad estandarizado para describir y evaluar la calidad de vida relacionada a la salud.
Aproximadamente el 63% de los pacientes que recibieron SIMPONI® al inicio de la extensión del estudio (semana 56) permanecieron en tratamiento hasta el final del estudio (la última administración de SIMPONI® en la semana 212).
Criterios de evaluación clínica durante el tratamiento de inducción: Los resultados en esta sección se basan en los pacientes aleatorizados durante el periodo de confirmación de la dosis en el Estudio 1 en CU.
Un porcentaje significativamente mayor de los pacientes del grupo de SIMPONI® alcanzó la respuesta clínica, la remisión clínica y la curación de la mucosa en comparación con el grupo del placebo en la semana 6. Además, los pacientes del grupo de SIMPONI® demostraron una mejoría significativamente mayor de la puntuación del IBDQ en comparación con el grupo del placebo.
Respuesta clínica: En el Estudio 1 en CU, la proporción de pacientes que alcanzaron la respuesta clínica hasta la semana 6 fue significativamente mayor en el grupo de SIMPONI® 200 mg/100 mg, el 51% en comparación con el grupo del placebo, 30.3%; p<0.0001 (Figura 11, Tabla 66)
Figura 11: Estudio 1 de CU: Porcentaje de los pacientes con CU que alcanzaron la respuesta clínica en la semana 6 en pacientes aleatorizados.
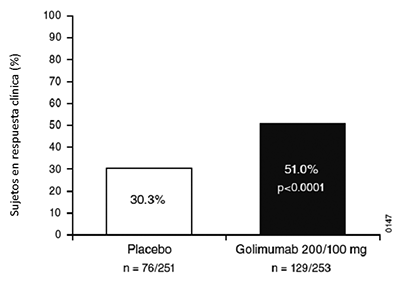
Remisión clínica: En el Estudio 1 en CU, una proporción significativamente mayor de los pacientes del grupo de SIMPONI® 200/100 mg estuvo en remisión clínica en la semana 6, 17.8% en comparación con el grupo del placebo, 6.4%; p<0.0001 (Figura 12, Tabla 66).
Figura 12: Estudio 1 de CU: Porcentaje de los pacientes con CU en remisión clínica en la semana 6 en pacientes aleatorizados.
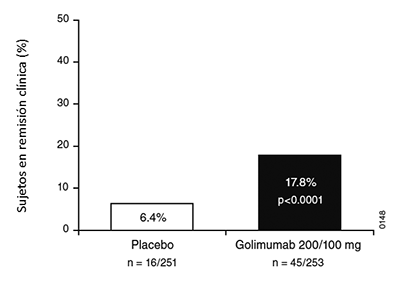
Curación de la mucosa: En el Estudio 1 en CU, una proporción significativamente mayor de los pacientes del grupo de SIMPONI® 200/100 mg alcanzaron la curación de la mucosa en la semana 6, 42.3% en comparación con el grupo del placebo, 28.7%; p<0.0014 (Tabla 66).
|
Tabla 66: Estudio 1 en CU: Proporción de los pacientes que alcanzaron respuesta clínica, remisión clínica y curación de la mucosa durante la inducción |
|||
|
Placebo (N = 251) |
SIMPONI® 200 mg /100 mg (N = 253) |
Valor-p |
|
|
Pacientes en respuestaa,d clínica en la semana 6 |
30.3% |
51.0% |
<0.0001 |
|
Pacientes en remisiónb,d clínica en la semana 6 |
6.4% |
17.8% |
<0.0001 |
|
Pacientes con curación de la mucosac,d en la semana 6 |
28.7% |
42.3% |
<0.0014 |
|
a Definido como una disminución desde el estado basal en la puntuación Mayo ≥ 30% y ≥ 3 puntos, acompañado por una disminución de la subpuntuación del sangrado rectal ≥ 1 o una subpuntuación del sangrado rectal de 0 o 1. b Definido como la puntuación Mayo ≤ 2 puntos, sin subpuntuación individual > 1. c Definido como 0 o 1 en la subpuntuación endoscópica de la puntuación Mayo. d Los pacientes que tuvieron un cambio prohibido en la medicación concomitante para CU, una ostomía o colectomía o un agente de estudio descontinuado debido a la ausencia del efecto terapéutico son considerados no estar en respuesta clínica, remisión clínica o curación de la mucosa desde el tiempo del evento en adelante. |
|||
Puntuación Mayo parcial: En el Estudio 1 en CU, una reducción mayor de la puntuación Mayo parcial fue evidente ya en la semana 2 en el grupo de SIMPONI® 200 mg/100 mg en comparación con el grupo del placebo y esta reducción se mantuvo hasta la semana 6.
Figura 13: Estudio 1 de CU: Mediana de la puntuación Mayo parcial hasta la semana 6 en pacientes aleatorizados
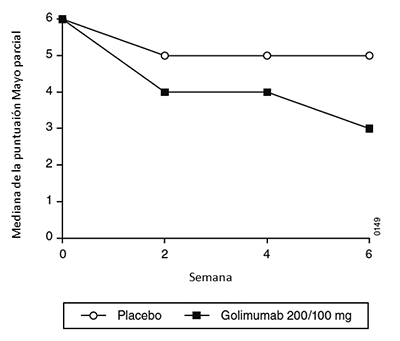
Mediciones de la calidad de vida relacionada con la salud: En el Estudio 1 en CU, la media de la mejoría de la puntuación del IBDQ en la semana 6 fue significativamente mayor en el grupo de SIMPONI® 200 mg/100 mg, 27.0 ± 33.72, en comparación con el grupo del placebo, 14.8 ± 31.25; p < 0.0001 (Tabla 67). Se observó una mejoría similar de la puntuación en todas las 4 dimensiones (es decir, intestinal, emocional, sistémico y social) del IBDQ (p ≤ 0.0005 en todas las comparaciones entre el grupo tratado con SIMPONI® 200 mg/100 mg y el grupo tratado con el placebo).
|
Tabla 67: Estudio 1 de CU: Mejoría de la puntuación del IBDQ |
|||
|
Placebo (N = 250) |
SIMPONI® 200/100 mg (N = 252) |
Valor-p |
|
|
Cambio desde el estado basal de la puntuación del IBDQa,b en la semana 6 (promedio ± SD) |
14.8 ± 31.25 |
27.0 ± 33.72 |
< 0.0001 |
|
a Cuestionario específico de la enfermedad intestinal inflamatoria b Los pacientes que tuvieron un cambio prohibido en la medicación concomitante para CU, una ostomía o colectomía o un agente de estudio descontinuado debido a la ausencia del efecto terapéutico previo a la visita de la semana 6 tuvieron un valor en el estado basal que se trasladó a la semana 6. |
|||
En la semana 6 en el Estudio 1 en CU, una proporción significativamente mayor de los pacientes en el grupo 200/100 mg presentaron: Una mejoría mayor a 20 puntos desde el estado basal en el IBDQ en comparación con el grupo del placebo en la semana 6 (50.6% frente a 35.5%, p=0006) y una mejoría en la dimensión del dolor/malestar del índice EQ-5D en comparación con el grupo del placebo (30.0% frente a 15.1%, p < 0.0001). Además, el incremento medio de la puntuación del resumen del componente físico de la encuesta de salud SF-36 y de la puntuación del resumen del componente mental del SF-36 fue significativamente mayor en el grupo de SIMPONI® 200/100 mg en comparación con el grupo del placebo (4.51 frente a 2.51, p=0.0009) y 4.57 frente a 1.60, p=0.0017; respectivamente).
Criterios de evaluación clínica durante el tratamiento de mantenimiento: Los resultados descritos en esta sección se basan en los pacientes que alcanzaron respuesta clínica con SIMPONI® desde la inducción previa con golimumab (N = 456).
La proporción de los pacientes que mantuvo la respuesta clínica hasta la semana 54 fue significativamente mayor en el grupo de SIMPONI® 100 mg en comparación con el grupo del placebo. Adicionalmente, la proporción de pacientes en respuesta clínica que alcanzaron la remisión clínica y curación de la mucosa en la semana 30 y la semana 54 fue significativamente mayor en el grupo de SIMPONI® 100 mg en comparación con el grupo del placebo.
Mantenimiento de la respuesta clínica hasta la semana 54: Se evaluó la actividad de la enfermedad CU mediante la puntuación Mayo parcial cada 4 semanas (menos de una respuesta fue confirmada por endoscopia). Por lo tanto, un paciente que mantuvo la respuesta de mantenimiento estuvo en un estado de respuesta clínica continua en cada evaluación hasta la semana 54. En el Estudio 2 en CU, la proporción de pacientes que mantuvo la respuesta clínica hasta la semana 54 fue significativamente mayor en el grupo de SIMPONI® 100 mg, 49.7% en comparación con el grupo del placebo, 31.2%; p<0.001 (Figura 14; Tabla 68).
Figura 14: Estudio 2 de CU: Porcentaje de los pacientes con CU que mantuvo la respuesta clínica hasta la semana 54 en pacientes aleatorizados
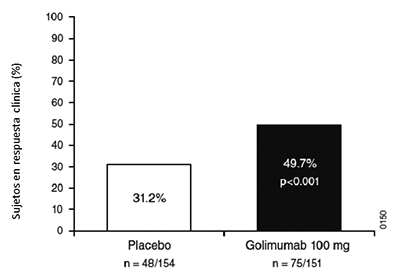
|
Tabla 68: Estudio 2 de CU: Respuesta clínica durante el tratamiento de mantenimiento |
|||
|
Placebo (N = 154) |
SIMPONI® 100 mg (N = 151) |
Valor-p |
|
|
Pacientes en respuesta clínicaa,b hasta la semana 54 |
31.2% |
49.7% |
< 0.001 |
|
a Definido como una disminución desde la semana 0 de inducción de la puntuación Mayo ≥ 30% y ≥ 3 puntos, acompañado de una disminución de la subpuntuación del sangrado rectal ≥ 1 o subpuntuación del sangrado rectal de 0 o 1. b Los pacientes que tuvieron un cambio prohibido en la medicación concomitante para CU, una ostomía o colectomía, un ajuste de dosis o un agente de estudio descontinuado debido a la ausencia del efecto terapéutico son considerados no estar en respuesta clínica desde el tiempo del evento en adelante. |
|||
Remisión clínica: Un paciente tenía que estar en remisión en la semana 30 y en la semana 54 (sin demostrar una pérdida de la respuesta en cualquier tiempo cronológico hasta la semana 54) para alcanzar la remisión duradera. En el Estudio 2 en CU, la proporción de los pacientes en remisión clínica en la semana 30 y en la semana 54 fue significativamente mayor en el grupo de SIMPONI® 100 mg, 27.8%, en comparación con el grupo del placebo, 15.6%, p = 0.004 (Figura 15; Tabla 69).
Figura 15: Estudio 2 de CU: Porcentaje de los pacientes con CU en remisión clínica en la semana 30 y en la semana 54 en pacientes aleatorizados
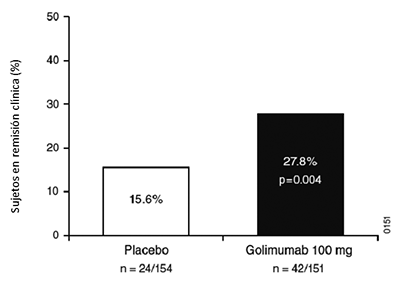
|
Tabla 69: Estudio 2 de CU: Remisión clínica durante el tratamiento de mantenimiento |
|||
|
Placebo (N = 154) |
SIMPONI® 100 mg (N = 151) |
Valor-p |
|
|
Pacientes en remisión clínicaa,b en la semana 30 y la semana 54 |
15.6% |
27.8% |
0.004 |
|
a Definido como una puntuación Mayo ≤ 2 puntos, sin subpuntuación individual > 1. b Los pacientes que tuvieron un cambio prohibido en la medicación concomitante para CU, una ostomía o colectomía, un ajuste de dosis o un agente de estudio descontinuado debido a la ausencia del efecto terapéutico son considerados no estar en remisión clínica desde el tiempo del evento en adelante. |
|||
En el Estudio 2 en CU, la proporción de pacientes en remisión clínica en la semana 54 fue mayor en el grupo de SIMPONI® 100 mg (33.8%), en comparación con el grupo del placebo (22.1%).
Curación de la mucosa: En el Estudio 2 en CU, la proporción de pacientes con curación de la mucosa en la semana 30 y en la semana 54 fue mayor en el grupo de 100 mg, 42.4%, en comparación con el grupo del placebo, 26.6%; p=0.002 (Tabla 70).
|
Tabla 70: Estudio 2 de CU: Curación de la mucosa durante el tratamiento de mantenimiento |
|||
|
Placebo (N = 154) |
SIMPONI® 100 mg (N = 151) |
Valor-p |
|
|
Pacientes con curación de la mucosaa,b en la semana 30 y la semana 54 |
26.6% |
42.4% |
0.002 |
|
a Definido como 0 o 1 en la puntuación endoscópica de la puntuación Mayo. b Los pacientes que tuvieron un cambio prohibido en la medicación concomitante para CU, una ostomía o colectomía, un ajuste de dosis o un agente de estudio descontinuado debido a la ausencia del efecto terapéutico son considerados no tener curación de la mucosa desde el tiempo del evento en adelante. |
|||
Entre los pacientes que ingresaron al Estudio 2 en CU con curación de la mucosa, la proporción de los pacientes que mantuvo la curación de la mucosa en la semana 30 y la semana 54 fue mayor en el grupo de 100 mg (44.6%), en comparación con el grupo del placebo (31.1%).
En el Estudio 2 en CU, la proporción de los pacientes con curación de la mucosa en la semana 54 fue mayor en el grupo de SIMPONI® 100 mg (46.4%), en comparación con el grupo del placebo (28.6%).
Mantenimiento de la remisión clínica: Entre los pacientes que ingresaron al Estudio 2 en CU en remisión clínica (35%, 160/456), la proporción de los pacientes que mantuvo la remisión clínica hasta la semana 54 fue mayor en el grupo de 100 mg (38.9%, 21/54) en comparación con el grupo del placebo (24.1%, 13/54; p = 0.098). Un análisis post hoc demostró que el tiempo de pérdida de la remisión clínica fue más largo en el grupo de SIMPONI® 100 mg en comparación con el grupo del placebo (la mediana del tiempo fue 50 semanas en comparación con 27 semanas, respectivamente).
Figura 16: Curva de Kaplan Meier para el tiempo hasta la pérdida de la remisión clínica en pacientes aleatorizados que estaban en remisión clínica en la semana 0 del Estudio 2 en CU.
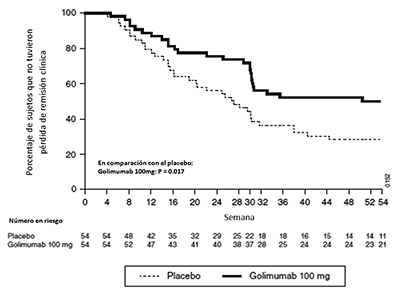
En el estudio 2 en CU, la proporción de los pacientes que mantuvo la remisión clínica hasta la semana 30 fue mayor en el grupo de SIMPONI® 100 mg (53.7%), en comparación con el grupo del placebo (33.3%).
Eliminación de los corticosteroides: En el estudio 2 en CU, entre los pacientes que recibieron concomitantemente corticosteroides en el estado basal (54%, 247/456), la proporción de los pacientes en remisión clínica y que no recibieron concomitantemente corticosteroides en la semana 54 fue similar en el grupo de 100 mg (23.2%, 19/82) en comparación con el grupo del placebo (18.4%, 16/87). Sin embargo, el análisis post hoc mostró la proporción de los pacientes que mantuvo la respuesta clínica hasta la semana 54 y que no recibieron concomitantemente corticosteroides en la semana 54 fue numéricamente mayor en el grupo de 100 mg (30.5%, 25/82) en comparación con el grupo del placebo (20.7%, 18/87). Además, la proporción de los pacientes que habían eliminado los corticosteroides en la semana 54 fue numéricamente mayor en el grupo de 100 mg (32.9%, 27/82) en comparación con el grupo del placebo (21.8%, 19/87). Entre los pacientes que ingresaron a la extensión del estudio, la proporción de sujetos que permaneció libre de corticosteroides se mantuvo generalmente hasta la semana 216.
Puntuación Mayo parcial hasta la semana 54: La mediana de la puntuación Mayo parcial en la semana 0 (del Estudio 2 en CU) se mantuvo en el grupo de SIMPONI® 100 mg hasta la semana 52 (con un incremento pequeño en la semana 54), la mediana de la puntuación Mayo parcial en el grupo del placebo se incrementó después de la semana 8 y continuó incrementándose en el tiempo hasta un valor en la semana 54 alcanzando el valor previo a la inducción con golimumab (Figura 17).
Figura 17: Estudio 2 de CU: Mediana de la puntuación Mayo parcial hasta la semana 54 en pacientes aleatorizados
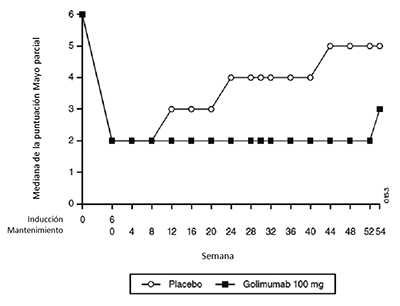
Subpuntuación Mayo: La proporción de pacientes en el Estudio 2 en CU que mantuvo la mejoría en cada subpuntuación Mayo desde la semana 0 hasta la semana 54 en el Estudio 2 en CU fue mayor en el grupo 100 mg en comparación con el grupo del placebo (Tabla 71).
Entre los pacientes que ingresaron a la extensión del estudio, la proporción de los sujetos con actividad de la enfermedad inactiva o leve evaluada por la subpuntuación de la evaluación general del médico de la puntuación Mayo fue generalmente sostenida hasta la semana 216.
|
Tabla 71: Proporción de los pacientes con subpuntuaciones Mayo que indican enfermedad inactiva o leve en la semana 0 de inducción hasta la semana 54 del estudio de mantenimiento en pacientes aleatorizados en el Estudio 2 en CU. |
||
|
Placebo N = 154 |
SIMPONI® 100 mg N = 151 |
|
|
Frecuencia de las deposiciones |
||
|
Semana 0 de la inducción |
11.7% |
15.9% |
|
Estudio de mantenimiento |
||
|
Semana 0 |
85.1% |
80.1% |
|
Semana 30 |
46.1% |
66.2% |
|
Semana 54 |
40.3% |
62.9% |
|
Sangrado rectal |
||
|
Semana 0 de la inducción |
44.8% |
37.7% |
|
Estudio de mantenimiento |
||
|
Semana 0 |
98.7% |
100.0% |
|
Semana 30 |
71.4% |
78.1% |
|
Semana 54 |
66.2% |
74.8% |
|
Evaluación general del médico |
||
|
Semana 0 de la inducción |
1.3% |
1.3% |
|
Estudio de mantenimiento |
||
|
Semana 0 |
83.1% |
83.4% |
|
Semana 30 |
44.2% |
65.6% |
|
Semana 54 |
39.0% |
57.6% |
|
Hallazgos endoscópicos |
||
|
Semana 0 de la inducción |
0% |
0% |
|
Estudio de mantenimiento |
||
|
Semana 0 |
70.1% |
74.2% |
|
Semana 30 |
38.3% |
60.3% |
|
Semana 54 |
34.4% |
55.0% |
Calidad de vida relacionada con la salud: Entre los pacientes con una mejoría mayor a 20 puntos en el IBDQ en el inicio del Estudio 2 en CU desde la semana 0 de un estudio de inducción, una mayor proporción de los pacientes en el grupo de SIMPONI® 100mg mantuvo la mejoría en el IBDQ hasta la semana 54, 40.0%, en comparación con el grupo del placebo, 27.8%; p=0.051.
INFORMACIÓN NO CLÍNICA:
Carcinogénesis, mutagénesis, deterioro de la fertilidad: No se han realizado estudios a largo plazo en animales con golimumab para evaluar el potencial carcinogénico o sus efectos en la fertilidad. Un estudio de la fertilidad realizado en ratones usando un anticuerpo análogo anti-TNFa del ratón no mostró deterioro de la fertilidad. No se han realizado estudios de mutagenicidad con golimumab.
ADVERTENCIAS Y PRECAUCIONES:
Infecciones: Se han notificado infecciones bacterianas (incluidas sepsis y neumonía), por micobacterias (tuberculosis), fúngicas invasivas y oportunistas, incluyendo casos fatales, en pacientes que recibían bloqueadores del TNF, incluyendo SIMPONI®. En los pacientes se ha presentado con frecuencia enfermedad diseminada en lugar de localizada. Algunas de estas infecciones graves se han producido en pacientes con terapia inmunosupresora concomitante que, junto con su enfermedad subyacente, podía predisponerlos a las infecciones.
Se deben considerar cuidadosamente los beneficios y los riesgos del tratamiento con SIMPONI® antes de iniciar o continuar dicho tratamiento en pacientes que hayan residido o viajado a regiones donde las infecciones fúngicas invasivas como histoplasmosis, coccidioidomicosis o blastomicosis son endémicas. En pacientes de riesgo tratados con SIMPONI® se debe sospechar una enfermedad fúngica invasiva si desarrollan una enfermedad sistémica grave. Pruebas de antígeno y anticuerpo pueden dar resultados negativos en algunos pacientes con infección activa. Se debe considerar un tratamiento antifúngico empírico adecuado mientras se realiza un estudio diagnóstico. Si es factible, la decisión de administrar un tratamiento antifúngico empírico se debe realizar consultando con un médico experto en el diagnóstico y el tratamiento de infecciones fúngicas invasivas y se debe tomar en consideración los riesgos de una infección fúngica grave y los riesgos de un tratamiento antifúngico.
SIMPONI® no debe administrarse en pacientes con infección activa clínicamente importante. Se debe tomar precaución cuando se considera el uso de SIMPONI® en pacientes con infección crónica o con antecedentes de infección recurrente. Se debe aconsejar a los pacientes que tomen las medidas adecuadas para evitar exponerse a factores potenciales de riesgo de infección.
Tuberculosis: Se deben evaluar en los pacientes los factores de riesgo de tuberculosis (incluyendo contacto cercano con alguna persona con tuberculosis activa) y determinar la existencia de infección tuberculosa latente antes del tratamiento con SIMPONI®. Se debe iniciar el tratamiento de la infección tuberculosa latente previo al tratamiento con SIMPONI®.
Se debe considerar el tratamiento antituberculoso antes de iniciar el tratamiento con SIMPONI® en pacientes con antecedentes de tuberculosis activa o latente, y en quienes no se pueda confirmar que hayan recibido un tratamiento adecuado.
Las pruebas para tuberculosis latente pueden dar resultados falso negativo, especialmente en pacientes inmunocomprometidos o gravemente enfermos. Antes de iniciar el tratamiento con SIMPONI®, se debe considerar el tratamiento para tuberculosis latente en pacientes que presentan factores de riesgo significativos de tuberculosis a pesar de los resultados negativos en las pruebas de tuberculosis latente. La decisión para iniciar el tratamiento antituberculoso en estos pacientes sólo se debe realizar después de consultar con un médico experto en el tratamiento de la tuberculosis y considerando el riesgo de infección tuberculosa latente y los riesgos del tratamiento antituberculoso.
En pacientes que reciben SIMPONI®, la tuberculosis se ha presentado frecuentemente como enfermedad diseminada o extrapulmonar. Ocurrieron casos de tuberculosis activa en pacientes tratados con SIMPONI® durante y después del tratamiento de tuberculosis latente. Se debe monitorear estrechamente los signos y síntomas de tuberculosis activa en los pacientes que reciben SIMPONI®, incluyendo pacientes que han dado resultado negativo en las pruebas de tuberculosis latente, pacientes con tratamiento de tuberculosis latente o pacientes tratados previamente de infección tuberculosa.
Neoplasias malignas: Se desconoce el papel potencial de la terapia con bloqueadores del TNF en el desarrollo de neoplasias malignas. Se debe tener precaución cuando se considera el tratamiento con bloqueadores del TNF en pacientes con antecedentes de neoplasias malignas, o cuando se considera continuar el tratamiento en los pacientes que desarrollan neoplasias malignas.
Neoplasias malignas pediátricas: Se han reportado casos posteriores a la comercialización, algunos fatales, de neoplasias malignas en niños, adolescentes y adultos jóvenes (hasta 22 años de edad) que recibían agentes bloqueadores del TNF (inicio del tratamiento ≤ 18 años de edad) para el tratamiento de Artritis Juvenil Idiopática (JIA), Enfermedad de Crohn u otras condiciones. Aproximadamente la mitad de los reportes fueron linfomas. Los otros casos representaban diversos tipos de neoplasias malignas, incluyendo neoplasias malignas que no son usualmente observados en niños y adolescentes. La mayoría de los pacientes recibían de forma concomitante inmunosupresores como metotrexato, azatioprina o 6-mercaptopurina. El papel de los bloqueadores del TNF en el desarrollo de neoplasias malignas en niños y adolescentes aún no es claro.
Linfoma: En el periodo controlado de los estudios clínicos de todos los agentes bloqueadores del TNF, incluyendo SIMPONI®, se han observado más casos de linfoma en los pacientes que recibieron tratamiento anti-TNF en comparación con los pacientes del grupo control. Durante la fase 2 y 3 de los estudios clínicos en AR, APs y EA con SIMPONI®, la incidencia de linfoma en los pacientes tratados con SIMPONI® fue mayor que la esperada en la población general. Los pacientes con artritis reumatoide y otras enfermedades inflamatorias crónicas, particularmente pacientes con enfermedad muy activa y/o con exposición crónica a tratamientos inmunosupresores, pueden tener mayor riesgo (varias veces mayor) que la población general de desarrollar linfoma, incluso en ausencia de tratamiento con bloqueadores del TNF.
Se han reportado casos raros de linfoma de células T hepatoesplénico (HSTCL) en pacientes tratados con otros agentes bloqueadores del TNF (ver sección Reacciones adversas). Este tipo raro de linfoma de células T tiene un curso de la enfermedad muy agresivo y es usualmente fatal. Casi todos estos casos han ocurrido en pacientes con Enfermedad de Crohn o con colitis ulcerativa. La mayoría fueron en varones adolescentes y adultos jóvenes. Casi todos estos pacientes habían recibido tratamiento con azatioprina (AZA) o 6-mercaptopurina (6–MP) concomitantemente con un bloqueador del TNF en o previo al diagnóstico. El riesgo potencial de la combinación de AZA o 6-MP y SIMPONI®, debe ser considerado cuidadosamente. No se puede excluir el riesgo del desarrollo de linfoma de células T hepatoesplénico en pacientes tratados con bloqueadores del TNF.
Leucemia: Se han reportado casos de leucemia aguda y crónica con el uso de bloqueadores del TNF, incluyendo SIMPONI®, en artritis reumatoide y otras indicaciones. Incluso en la ausencia del tratamiento con bloqueadores del TNF, los pacientes con artritis reumatoide pueden tener mayor riesgo (aproximadamente el doble) que la población general de desarrollar leucemia.
Neoplasias malignas distintas al linfoma: En el periodo controlado de los estudios clínicos de fase 2 y fase 3 en AR, APs, EA y CU con SIMPONI®, la incidencia de neoplasias malignas distintas al linfoma (excluyendo el cáncer de piel no melanoma) fue similar entre los grupos con SIMPONI® y el control.
En un ensayo clínico exploratorio donde se evaluó el uso de SIMPONI® en pacientes con asma persistente severo, más pacientes tratados con SIMPONI® reportaron neoplasias malignas comparado con los pacientes del grupo control (ver sección Reacciones Adversas). Se desconoce la significancia de estos hallazgos.
Displasia/Carcinoma de colon: Se desconoce si el tratamiento con SIMPONI® tiene influencia sobre el riesgo de desarrollar displasia o cáncer de colon. Todos los pacientes con colitis ulcerativa que presentan un mayor riesgo de displasia o carcinoma de colon (por ejemplo, pacientes con colitis ulcerativa de larga evolución o colangitis esclerosante primaria), o quienes tuvieron un antecedente de displasia o carcinoma de colon deben someterse a una revisión a intervalos regulares para el diagnóstico de displasia antes de recibir el tratamiento, y a lo largo del curso de su enfermedad. Esta evaluación debe incluir colonoscopia y biopsia según recomendaciones locales. En pacientes con displasia de nuevo diagnóstico, tratados con SIMPONI®, se debe revisar cuidadosamente los riesgos y los beneficios para los pacientes y se debe considerar la continuación del tratamiento.
Cánceres de piel: Se ha reportado melanoma y carcinoma de células de Merkel en pacientes tratados con agentes bloqueadores del TNF, incluyendo SIMPONI® (ver sección Reacciones Adversas). Se recomienda exámenes periódicos de la piel para todos los pacientes, especialmente para aquellos con factores de riesgo para cáncer de piel.
Reactivación del virus de la hepatitis B: Tal y como ocurre con otros medicamentos inmunosupresores, el uso de los agentes bloqueadores del TNF, incluyendo SIMPONI® se ha relacionado con la reactivación del virus de la hepatitis B (HBV) en pacientes portadores crónicos del virus (es decir, positivos para el antígeno de superficie). Los pacientes deben hacerse la prueba de infección por HBV antes de iniciar el tratamiento con los inmunosupresores, incluyendo SIMPONI®. Para aquellos pacientes que dan resultado positivo para el antígeno de superficie de la hepatitis B, se recomienda consultar con un médico experto en el tratamiento de la hepatitis B. Se debe evaluar y monitorear adecuadamente a los portadores crónicos de la hepatitis B antes de iniciar el tratamiento, durante el tratamiento y durante varios meses después de la descontinuación del tratamiento con SIMPONI®.
Insuficiencia cardiaca congestiva (ICC): Se han reportado casos de empeoramiento de la insuficiencia cardiaca congestiva (ICC) y casos de nueva aparición de ICC con bloqueadores del TNF, incluyendo SIMPONI®. Algunos casos tuvieron un desenlace mortal. No se ha estudiado SIMPONI® en pacientes con ICC. SIMPONI® debe ser usado con precaución en pacientes con insuficiencia cardiaca. Si se decide administrar SIMPONI® a pacientes con insuficiencia cardiaca, estos deben ser estrechamente monitoreados durante el tratamiento, y se debe descontinuar el tratamiento con SIMPONI® si aparecen síntomas nuevos o se observa el empeoramiento de los síntomas de la insuficiencia cardiaca.
Trastornos desmielinizantes: El uso de agentes bloqueadores del TNF se ha relacionado con casos de nueva aparición o exacerbación de los síntomas clínicos y/o evidencia radiográfica de trastornos desmielinizantes del sistema nervioso central, incluyendo esclerosis múltiple (EM) y trastornos desmielinizantes periféricos, incluyendo síndrome de Guillain-Barré. Los prescriptores deben tener precaución al considerar el uso de los bloqueadores del TNF, incluyendo SIMPONI®, en pacientes con trastornos desmielinizantes del sistema nervioso central o periférico. Se debe considerar la descontinuación del tratamiento con SIMPONI® si se presentan estos trastornos.
Procesos autoinmunes: El tratamiento con bloqueadores del TNF, incluyendo SIMPONI®, puede resultar en la formación de anticuerpos antinucleares (ANA) y, raramente, en el desarrollo de un síndrome tipo lupus (ver sección Reacciones Adversas). Si un paciente desarrolla síntomas indicativos de un síndrome tipo lupus después del tratamiento con SIMPONI® se debe descontinuar el tratamiento.
Administración concomitante de SIMPONI® con anakinra: En estudios clínicos con el uso concomitante de anakinra y otro agente bloqueador del TNF, etanercept, se han observado infecciones graves y neutropenia, sin beneficio clínico añadido. Debido a la naturaleza de las reacciones adversas observadas con este tratamiento combinado, la aparición de toxicidades similares puede resultar también de la combinación de anakinra con otros agentes bloqueadores del TNF. Por lo tanto, no se recomienda combinar SIMPONI® con anakinra.
Administración concomitante de SIMPONI® con abatacept: En estudios clínicos, la administración concomitante de agentes bloqueadores del TNF y abatacept se ha relacionado con un mayor riesgo de infecciones incluyendo infecciones graves en comparación con la administración sola de los agentes bloqueadores del TNF, sin beneficio clínico añadido. Debido a la naturaleza de las reacciones adversas observadas con el tratamiento combinado de agentes bloqueadores del TNF y abatacept, no se recomienda la combinación de SIMPONI® con abatacept.
Administración concomitante con otras terapias biológicas: No hay información suficiente relativa al uso concomitante de SIMPONI® con otras terapias biológicas utilizadas para tratar las mismas afecciones que SIMPONI®. No se recomienda el uso concomitante de SIMPONI® con estos medicamentos biológicos debido a la posibilidad de un incremento del riesgo de infección.
Cambio entre terapias biológicas: Cuando se cambia de una terapia biológica a otra, se debe continuar monitoreando a los pacientes, ya que la superposición de la actividad biológica podría incrementar el riesgo de infección.
Reacciones hematológicas: Se han reportado casos de pancitopenia, leucopenia, neutropenia, agranulocitosis y trombocitopenia en pacientes que recibían bloqueadores del TNF, incluyendo SIMPONI®. Se debe tener precaución en los pacientes tratados con SIMPONI® que tengan citopenias significativas o hayan tenido antecedentes de citopenias significativas.
Vacunas de microorganismos vivos/Agentes infecciosos terapéuticos: Los pacientes tratados con SIMPONI® pueden recibir vacunas concomitantemente, a excepción de las vacunas de microorganismos vivos. En pacientes que están recibiendo terapia anti-TNF, los datos disponibles sobre la respuesta a la vacunación con vacunas de microorganismos vivos o sobre la transmisión secundaria de la infección por vacunas de microorganismos vivos es limitada. El uso de vacunas de microorganismos vivos puede causar infecciones clínicas, incluyendo infecciones diseminadas.
Otros usos de los agentes infecciosos terapéuticos tales como bacterias vivas atenuadas (por ejemplo, la instilación BCG en la vejiga para el tratamiento de cáncer) podrían causar infecciones clínicas, incluyendo infecciones diseminadas. Se recomienda que no se administren los agentes infecciosos terapéuticos de forma concomitante con SIMPONI®.
Vacunas no-vivas: Los pacientes con artritis psoriásica tratados con SIMPONI® en un estudio de fase 3 en APs consiguieron desarrollar una respuesta inmune efectiva de las células B frente a la vacuna antineumocócica polisacárida. Un número similar de pacientes con artritis psoriásica que recibieron SIMPONI® y los que no recibieron SIMPONI® tuvieron por lo menos un incremento de 2 veces la valoración de los anticuerpos. La proporción de pacientes con respuesta a la vacuna neumocócica fue menor en los pacientes tratados con SIMPONI® y en los pacientes control que recibieron MTX en comparación con los pacientes que no recibieron MTX. En general, los datos indican que SIMPONI® no suprime la respuesta inmune humoral a esta vacuna.
Reacciones alérgicas:
Sensibilidad al látex: La funda de la aguja de la jeringa precargada que viene en la pluma autoinyectable/precargada contiene goma seca natural (un derivado del látex), el cual puede causar reacciones alérgicas a las personas sensibles al látex.
Reacciones de hipersensibilidad: Durante la experiencia posterior a la comercialización, se han reportado reacciones graves de hipersensibilidad sistémica (incluida reacción anafiláctica), después de la administración de SIMPONI®. Algunas de estas reacciones ocurrieron después de la primera administración de SIMPONI®. Si ocurre una reacción anafiláctica u otra reacción grave, se debe interrumpir inmediatamente la administración de SIMPONI® y se debe iniciar un tratamiento adecuado.
Poblaciones especiales:
Uso geriátrico: En estudios de fase 3 por vía subcutánea en AR, APs y EA y en estudios de fase 3 por vía intravenosa en AR, no se observaron diferencias generales referentes a las reacciones adversas, reacciones adversas graves e infecciones graves en pacientes de 65 años o mayores que recibieron SIMPONI® en comparación con los pacientes más jóvenes. En estudios de fase 3 por vía intravenosa en APs y EA hubo un número de pacientes de 65 años o mayores para determinar si respondían de manera diferente que los pacientes de 18 y 65 años de edad. En CU, hubo un número insuficiente de pacientes de 65 años o mayores para determinar si ellos responden de forma diferente que los pacientes de 18 a 65 años. Debido a que la incidencia de infecciones es mayor en la población de edad avanzada en general, se debe tener precaución al momento de tratar a los ancianos. No hubo pacientes de 65 años o mayores en el estudio de nr Axial SpA.
PRECAUCIONES ESPECIALES DE CONSERVACIÓN: Conservar en nevera entre 2 ºC y 8 ºC
Conservar en el embalaje original hasta su uso.
Proteger de la luz.
No congelar.
No agitar.
Mantener fuera del alcance de los niños.
POSOLOGÍA Y ADMINISTRACIÓN: SIMPONI® se administra mediante inyección subcutánea y SIMPONI® IV se administra mediante infusión intravenosa.
No se ha establecido la eficacia y seguridad del cambio entre las formulaciones de administración intravenosa y subcutánea.
Dosis – Adultos:
Artritis reumatoide:
Inyección subcutánea: 50 mg de SIMPONI® administrado mediante inyección subcutánea una vez al mes, en el mismo día de cada mes.
Infusión intravenosa: 2 mg/kg de SIMPONI® IV administrado mediante infusión intravenosa de 30 minutos en la semana 0 y 4 y posteriormente cada 8 semanas (ver sección Instrucciones de uso, manejo y disposición).
Artritis psoriásica:
Inyección subcutánea: 50 mg de SIMPONI® administrado mediante inyección subcutánea una vez al mes, el mismo día de cada mes.
Infusión intravenosa: 2 mg/kg de SIMPONI® IV administrado mediante infusión intravenosa de 30 minutos en la semana 0 y 4 y posteriormente cada 8 semanas (ver sección Instrucciones de uso, manejo y disposición).
Espondilitis anquilosante:
Inyección subcutánea: 50 mg de SIMPONI® administrado mediante inyección subcutánea una vez al mes, el mismo día de cada mes.
Infusión intravenosa: 2 mg/kg de SIMPONI® IV administrado mediante infusión intravenosa de 30 minutos en la semana 0 y 4 y posteriormente cada 8 semanas (ver sección Instrucciones de uso, manejo y disposición).
Espondiloartritis axial no radiográfica: 50 mg de SIMPONI® administrado mediante inyección subcutánea una vez al mes, el mismo día de cada mes.
Colitis ulcerativa:
Pacientes con peso corporal menor de 80 Kg: Administrar inicialmente 200 mg de SIMPONI®, mediante administración subcutánea, seguidos por 100 mg en la semana 2 y posteriormente 50 mg cada 4 semanas (ver sección Propiedades farmacodinámicas).
Pacientes con peso corporal mayor de 80 Kg: 200 mg de SIMPONI®, mediante administración subcutánea en la semana 0, seguido por 100 mg en la semana 2, y posteriormente 100 mg cada 4 semanas (ver sección Propiedades farmacodinámicas).
Durante el tratamiento de mantenimiento, los corticosteroides se pueden reducir de acuerdo con las guías de la práctica clínica.
Los datos disponibles sugieren que la respuesta clínica se alcanza generalmente dentro de la semana 12 a la 14 de tratamiento (después de 4 dosis). La continuación del tratamiento debe ser reconsiderada en pacientes que no muestran evidencia de beneficio terapéutico dentro de este período de tiempo.
MÉTODO DE ADMINISTRACIÓN: SIMPONI® y SIMPONI® IV deben utilizarse bajo la dirección y supervisión de un médico.
Inyección subcutánea: Después de recibir entrenamiento adecuado en la técnica de inyección subcutánea, el paciente puede autoinyectarse SIMPONI®, si el médico determina que es lo adecuado y con el seguimiento médico necesario.
Al momento de la administración, si se requieren múltiples inyecciones, las inyecciones deben ser administradas en diferentes partes del cuerpo (ver Instrucciones para Inyectar SIMPONI® con la pluma autoinyectable/precargada de uso único SmartJect®). Las instrucciones completas para la administración de SIMPONI® se proporcionan en las secciones “Instrucciones de uso, manejo y disposición” y la sección “Instrucciones para Inyectar SIMPONI® con la pluma autoinyectable/precargada de uso único SmartJect®”. Se debe enseñar a los pacientes a inyectarse la cantidad completa de SIMPONI® según las instrucciones descritas en la sección “Instrucciones para Inyectar SIMPONI® con la pluma autoinyectable/precargada de uso único SmartJect®”.
Infusión intravenosa: La infusión intravenosa de SIMPONI® IV debe ser administrada por profesionales de la salud cuidadosamente entrenados para detectar cualquier evento relacionado con la infusión.
Las instrucciones completas para la infusión intravenosa de SIMPONI® se proporciona en la sección “Instruciones de uso, manejo y disposición”.
Poblaciones especiales:
Pediátricos (17 años de edad y menores): La seguridad y la eficacia de SIMPONI® no han sido establecidas en pacientes pediátricos de 17 años y menores; por lo tanto, no se pueden hacer recomendaciones sobre la dosificación (ver sección Propiedades farmacodinámicas).
Personas mayores (65 años de edad y mayores): No se requiere ajuste de la dosis para los pacientes de edad avanzada (ver sección Advertencias y precauciones).
Insuficiencia renal y hepática: No se han realizado estudios específicos con SIMPONI® en pacientes con insuficiencia renal e insuficiencia hepática.
SOBREDOSIS: En un estudio clínico se administraron dosis únicas por vía intravenosa de hasta 10 mg/kg sin toxicidad limitante de la dosis. En caso de sobredosis, se recomienda monitorear al paciente para detectar cualquier signo o síntoma de efectos adversos e iniciar inmediatamente un tratamiento sintomático adecuado.
PRESENTACIÓN: SIMPONI® Solución inyectable 50 mg (Reg. San. INVIMA 2018 M-0012615-R1). SIMPONI® IV (Reg. San. INVIMA 2015 M-0016181).
JANSSEN-CILAG, S. A.
Av. Calle 26 No. 69-76 - Edificio Elemento
Torre 2, Piso 11 - PBX: +57 1 927 1200 - Bogotá, D. C.
NATURALEZA Y CONTENIDO DEL ENVASE:
Pluma autoinyectable/precargada: SIMPONI® se presenta como solución estéril de uso único, en una jeringa de vidrio tipo 1 con una aguja fija de acero inoxidable. La jeringa se encuentra dentro de una pluma autoinyectable precargada de uso único. La jeringa va tapada con un tapón recubierto y la aguja cubierta con una funda, para evitar la pérdida de solución a través de la aguja y para protegerla durante la manipulación previa a la administración. La aguja fija es una aguja de acero inoxidable 5-biselada 27G de media pulgada. Las fundas de las agujas se fabrican utilizando goma natural seca, la cual contiene látex (ver sección Advertencias y precauciones).
SIMPONI® no contiene conservantes. La solución es de trasparente a ligeramente opalescente y de incoloro a ligeramente amarillo, con un pH de aproximadamente 5.5. Cada ml de SIMPONI® contiene 100 mg de golimumab, 0.87 mg de L-histidina y L-histidina hidrocloruro, 41.0 mg de sorbitol, 0.15 mg de polisorbato 80 y agua para inyección. Hay sólo una concentración de SIMPONI® disponible, 50 mg de golimumab en 0.5 ml.
SIMPONI® está disponible en envases con 1 pluma autoinyectable precargada y en envases con 3 plumas autoinyectables precargadas. Puede que no se comercialicen todos los tamaños de envase.
SIMPONI® se envasa en una caja exterior no reutilizable.
Vial: SIMPONI® IV para infusión intravenosa se presenta en viales de uso único.
SIMPONI® IV no contiene conservantes. La solución es de incoloro a ligeramente amarillo, con un pH de aproximadamente 5.5. Cada 4 ml del vial de SIMPONI® IV contiene 50 mg de golimumab, 1.14 mg de L-histidina y 6.42 mg de L-histidina hidrocloruro, 180 mg de sorbitol, 0.6mg de polisorbato 80 y agua para inyección.
SIMPONI® IV se envasa en una caja exterior no reutilizable.
Instrucciones de uso, manejo y disposición:
Pluma autoinyectable/precargada: Ver sección Instrucciones para Inyectar SIMPONI® con la pluma autoinyectable/precargada de uso único SmartJect.
Vial: Usar técnica aséptica:
1. Calcular la dosis y el número de viales de SIMPONI® IV necesarios basados en el peso del paciente. Cada vial de 4 ml de SIMPONI® IV contiene 50 mg de golimumab.
2. Revisar que la solución sea de incolora a color amarillo pálido. La solución puede tener partículas translúcidas finas, ya que golimumab es una proteína. No utilizar si se observan partículas opacas, decoloración u otras partículas extrañas.
3. Diluir el volumen total de la dosis de la solución de SIMPONI® IV en 100 ml con cloruro de sodio 0.9% p/v para perfusión. Esto se puede lograr retirando un volumen de la solución de cloruro de sodio 0.9% p/v de la botella de vidrio de 100 ml o de bolsa de infusión igual al volumen de SIMPONI® IV y desechar la solución retirada. Alternativamente, SIMPONI® I.V. se puede diluir usando el mismo método descrito anteriormente con 0.45% de cloruro de sodio p/v para infusión.
4. Agregue lentamente el volumen total de la solución de SIMPONI® IV a la botella o bolsa de infusión de 100 ml. Mezclar con cuidado.
5. Inspeccionar visualmente para detectar partículas extrañas o decoloración antes de su administración. No utilizar si se observan partículas opacas visiblemente, decoloración o partículas extrañas.
6. Administrar la solución diluida por un período de 30 ± 10 minutos. La infusión de la solución diluida debe ser completado dentro de las 6 horas después de la preparación.
7. Use solo un equipo de infusión de una línea, no pirógena, estéril, con filtro de baja unión a proteínas (tamaño de poro de 0.22 micrómetros o menos). No conservar la porción no utilizada de la solución de infusión para reutilizarla.
8. No se han realizado estudios de compatibilidad físico bioquímica para evaluar la administración conjunta de SIMPONI® IV con otros agentes. No administrar SIMPONI® IV concomitantemente en la misma línea intravenosa con otros agentes.
9. Cualquier material utilizado o los residuos deben ser eliminados de acuerdo con las exigencias locales.
Instrucciones para Inyectar SIMPONI® con la pluma autoinyectable/precargada de uso único SmartJect®: Si desea autoinyectarse SIMPONI®, un profesional de la salud deberá enseñarle a preparar la inyección y administrársela a sí mismo. Si no le han enseñado, contacte a su profesional de la salud para acordar una sesión de entrenamiento.
Paso 1: Preparse para utilizar el autoinyectable: El siguiente diagrama muestra cómo es el autoinyectable:
|
English |
Español |
|
Cap |
Tapa |
|
Security seal |
Precinto de seguridad |
|
Grenn Safety sleeve |
Manguito verde seguridad |
|
Raised Part of Button |
Parte elevada del Botón |
|
Expiration date |
Fecha de caducidad |
|
Viewing window |
Visor |
|
Clear cover |
Cubierta transparente |
NO agitar el autoinyectable en ningún momento.
NO retirar la tapa del autoinyectable hasta inmediatamente antes de la inyección.
Revisar la fecha de caducidad:
• Revisar la fecha de caducidad (indicada como “EXP”) en el autoinyectable.
• También puede ver la fecha de caducidad en el envase externo de cartón.
• Si ya ha pasado la fecha de caducidad, NO utilice el autoinyectable y contacte con su médico, farmacéutico o con el distribuidor local de este medicamento para pedir asistencia.
Revisar el precinto de seguridad:
• Revisar el precinto de seguridad que rodea la tapa del autoinyectable. Si el precinto de seguridad está roto, NO use el autoinyectable y contacte con su médico, farmacéutico o con el distribuidor local de este medicamento para pedir asistencia.
Esperar 30 minutos:
• Para asegurar la correcta inyección, deje el autoinyectable fuera de su caja, a temperatura ambiente y fuera del alcance de los niños, durante 30 minutos.

|
English |
Español |
|
Wait 30 minutes |
Esperar 30 minutos |
NO calentar el autoinyectable de ninguna otra forma a la indicada en el punto anterior (por ejemplo, NO lo calentar en el microondas o en agua caliente).
NO retirar la tapa del autoinyectable mientras espera que se alcance la temperatura ambiente.
Preparar el resto de los elementos:
• Preparar el resto de los elementos necesarios para su inyección. Estos incluyen un hisopo con alcohol, un trozo de algodón o gasa y un contenedor para objetos cortantes.
Revisar el líquido en el autoinyectable:
• Observar a través del visor para asegurarse de que el líquido del autoinyectable es transparente a ligeramente opalescente y de incoloro a ligeramente amarillo.
• También puede observar una burbuja de aire. Esto es normal.
NO lo utilice si el líquido presenta decoloración (alteración del color), está turbio o contiene partículas. En ese caso, contactar a su médico, farmacéutico o con el distribuidor local de este medicamento para pedir asistencia.
Paso 2: Elegir y preparar el lugar de inyección:
Elegir el lugar de inyección:
• El lugar de inyección recomendado es la parte media de la cara anterior de los muslos.
• También puede usar la parte inferior del abdomen, debajo del ombligo, a excepción de la zona correspondiente a dos pulgadas (5.1 cm) directamente debajo del ombligo.
• Si la inyección es aplicada por un cuidador, éste puede utilizar también la cara externa de los brazos.
• Si son necesarias múltiples inyecciones para una única administración, las inyecciones se administrarán en diferentes zonas del cuerpo.
NO inyectar en zonas donde la piel esté sensible, con hematomas, roja, descamada o dura. Evitar zonas con cicatrices o estrías.
Preparar el lugar de inyección:
• Lavar sus manos cuidadosamente con agua caliente y jabón.
• Limpiar el sitio de inyección con un hisopo con alcohol.
NO tocar nuevamente esta zona antes de aplicar la inyección. Dejar secar la piel antes de inyectar.
NO abanique ni sople sobre la zona limpia.
Paso 3: Inyectar la medicación:
Retirar la tapa:
NO retirar la tapa hasta cuando esté preparado para inyectar la medicación. La medicación debe ser inyectada dentro de los 5 minutos después que la tapa ha sido retirada.
• Cuando esté listo para la inyección, gire suavemente la tapa para romper el precinto de seguridad.
• Desprenda la tapa tirando de ella y colocarla inmediatamente en la basura.
NO volver a colocar la tapa porque puede dañar la aguja que está dentro del autoinyectable.
Nota: NO utilizar el autoinyectable si se ha caído sin la tapa. Si el autoinyectable se le cae sin la tapa, contacte a su médico, farmacéutico o con el distribuidor local de este medicamento para pedir asistencia.
Empujar el autoinyectable contra la piel:
• Sostener el autoinyectable en su mano con comodidad. NO presionar el botón en este momento.
• Empujar el extremo abierto del autoinyectable firmemente contra la piel en un ángulo de 90 grados.
• NO presionar el botón hasta que el autoinyectable este apretado firmemente contra la piel y el maguito de seguridad se deslice dentro de la cubierta transparente.
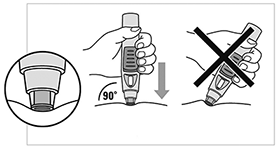
• Se recomienda la inyección sin pellizcar la piel (Figura de la izquierda). Sin embargo, si prefiere, puede pellizcar la piel para crear una superficie más firme para la inyección (Figura de la derecha).
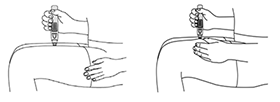
Presionar el botón para inyectar
• Continúe sosteniendo firmemente el autoinyectable contra la piel, y presionar la parte frontal elevada del botón con los dedos o con el pulgar. No será capaz de presionar el botón a menos que autoinyectable esté firmemente apretada contra la piel y el manguito de seguridad se deslice dentro de la cubierta transparente.
• Una vez que el botón ha sido presionado, el botón permanecerá presionado de modo que no necesita seguir presionando.
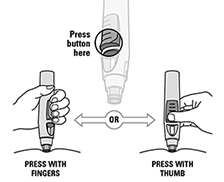
|
English |
Español |
|
Press button here |
Presionar el botón aqui |
|
Press with fingers |
Presionar con los dedos |
|
Press with thumb |
Presionar con el pulgar |
• Oirá un sonido fuerte, un ‘clic’ - no se alarme. El primer ‘clic’ indica que la aguja se ha insertado y ha empezado la inyección. Usted puede sentir o no el pinchazo en ese momento.
NO levantar el autoinyectable de la piel. Si retira el autoinyectable de la piel, puede no recibir toda la dosis del medicamento.
Esperar por el segundo “clic”:
• Continúe sosteniendo firmemente el autoinyectable contra la piel hasta que oiga un segundo “clic” (puede tardar alrededor de 3 a 6 segundos, pero puede pasar hasta 15 segundos para que oiga el segundo “clic”).
• El segundo “clic” indica que la inyección ha terminado y que la aguja se ha retraído dentro del autoinyectable. Nota: Si tiene problemas auditivos y no oye el segundo “click”, cuente 15 segundos desde el momento en que presiono el botón y luego levante el autoinyectable del lugar de inyección.
• Levante el autoinyectable del lugar de inyección.
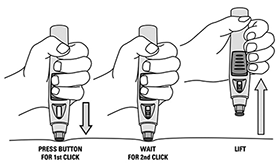
|
English |
Español |
|
Press button for 1st Click |
Presionar el botón hasta oir el primer click |
|
Wait for 2nd Click |
Esperar hasta oir el segundo click |
|
Lift |
Levantar el autoinyectable |
Paso 4: Después de la inyección:
Revisar el visor:
• Después de inyectar, revisar el visor para asegurarse de que el indicador amarillo está visible.
• El indicador amarillo puede no llenar completamente el visor. Esto es normal.
• Esto indica que el autoinyectable ha funcionado correctamente.
• Si piensa que no ha recibido su inyección, revisar nuevamente el indicador amarillo para confirmar que se haya administrado la dosis.
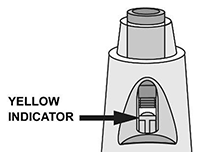
|
English |
Español |
|
Yellow indicator |
Indicador amarillo |
• Si el indicador amarillo no está visible en el visor, contacte con su médico, farmacéutico o con el distribuidor local de este medicamento para pedir asistencia. NO se administre una segunda dosis sin hablar con su médico.
Desechar el autoinyectable:
• Desechar inmediatamente el autoinyectable en un contenedor para objetos cortantes.
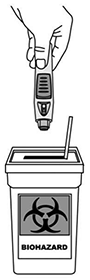
|
English |
Español |
|
Biohazard |
Peligro Biológico |
• Desechar el contenedor para objetos cortantes de acuerdo con la normativa local cuando el contenedor está lleno.
Utilizar un trozo de algodón o gasa:
• Puede haber una pequeña cantidad de sangre o líquido en el lugar de inyección, lo cual es normal.
• Puede presionar un trozo de algodón o gasa sobre el lugar de la inyección y mantenerlo durante 10 segundos.
NO frotar el lugar de inyección.
• Puede cubrir el lugar de inyección con un pequeño vendaje adhesivo, si es necesario.


