

TEMODAL
TEMOZOLOMIDA
Vial
Vial(es) , Polvo liofilizado
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
FARMACOLOGÍA CLÍNICA Y FARMARCOCINÉTICA:
Absorción: Después de la administración oral a pacientes adultos, TEMODAL® se absorbe rápida y completamente con concentraciones máximas alcanzadas tan solo 20 minutos después de la dosis, (tiempos entre 0,5 y 1,5 horas). En un ensayo con temozolomida radiomarcada con C14, la excreción fecal media de C14 durante los 7 días después de la dosis fue de 0,8% que indica una absorción completa.
Distribución: Las concentraciones plasmáticas de temozolomida aumentan de forma relacionada con la dosis. El volumen aparente de distribución medio en adultos y en pacientes pediátricos después de una sola dosis de TEMODAL® osciló entre 0,35 a 0,63 L/Kg y 0,35 a 0,41 L/Kg, respectivamente. La depuración plasmática, el volumen de distribución y la vida media son independientes de la dosis, tienen muy bajos coeficientes de variación, son predecibles y reproducibles. Temozolomida se elimina rápidamente y no se acumula en el plasma después de dosis múltiples diarias. Los pacientes pediátricos presentaron un ABC mayor para una dosis comparable (por área de superficie corporal en m2) que los pacientes adultos.
TEMODAL® demuestra baja unión a proteínas (12% a 16%), y por lo tanto no se espera que interactúe con agentes altamente unidos a proteínas.
Metabolismo y eliminación: Las principales vías de eliminación plasmáticas de TEMODAL® fueron hidrólisis no enzimática a MTIC y la excreción renal del fármaco original. Después de la administración oral, aproximadamente el 5% de la dosis se recupera sin cambios en la orina durante 24 horas, y el resto del C14 recuperado se excreta en forma de 5-aminoimidazol-4-carboxamida (AIC), el ácido carboxílico temozolomida (TMA, por sus siglas en inglés) o metabolitos polares no identificados.
TMA (temozolomida ácido carboxílico) fue el único metabolito de importancia y la excreción urinaria de TMA representaba menos del 3% de la dosis de TEMODAL®. El metabolismo mediado por el citocromo P450 (CYP450) tal como se evaluó mediante la medición de los niveles de TMA no contribuye significativamente a la depuración plasmática de TEMODAL®, por lo tanto la depuración de TEMODAL® no debería verse afectada en un grado clínicamente significativo por la interacción de medicamentos, ni debería la administración de TEMODAL® alterar el metabolismo de otros fármacos.
Análisis de población basados en datos farmacocinéticos de TEMODAL® revelaron que la depuración plasmática de TEMODAL® era independiente de la edad, función renal, función hepática, uso de tabaco o el uso de dexametasona, proclorperazina, fenitoina, carbamazepina, ondansetron, antagonistas del receptor H2, o fenobarbital. La depuración de TEMODAL® estuvo relacionada significativamente al tamaño corporal y más estrechamente relacionada al área de superficie corporal. Las mujeres tuvieron una depuración más baja estadísticamente significativa, pero no clínicamente significativa que los pacientes hombres.
En un estudio abierto, cruzado de dos vías de la farmacocinética de temozolomida oral e intravenosa en pacientes con malignidades primarias del SNC, TEMODAL® polvo para solución administrado por infusión durante 90 minutos demostró ser bioequivalente para Cmáx y ABC de temozolomida y MTIC en comparación con TEMODAL® cápsulas orales, tras administración de una dosis de 150 mg/m2. Los valores de Cmáx promedio para temozolomida y MTIC fueron 7.4 µg/mL y 320 ng/mL, respectivamente, tras infusión intravenosa de 90 minutos. Los valores promedio ABC para temozolomida y MTIC fueron 25 µg•hr/mL y 1,004 ng•hr /mL, respectivamente.
INDICACIONES Y USO:
TEMODAL® está indicado para el tratamiento de pacientes con:
• Glioblastoma Multiforme recién diagnosticado concomitantemente con radioterapia, y luego como tratamiento coadyuvante.
• Gliomas malignos, tales como Glioblastoma Multiforme o Astrocitoma Anaplásico, que presenten recurrencia o progresión después de la terapia estándar.
• Melanoma maligno metastásico avanzado, como primera línea de tratamiento.
CONTRAINDICACIONES: TEMODAL® está contraindicado en pacientes con historia de hipersensibilidad a sus componentes o a la dacarbazina1.
1 Ya que ambos son metabolizados al MTIC (5-(3-metiltriacen-1-il) imidazol-4-carboxamida)).
TEMODAL® está contraindicado en el embarazo y lactancia (ver Uso durante el embarazo y el período de lactancia).
El uso de TEMODAL® está contraindicado en pacientes con mielosupresión severa.
REACCIONES Y EVENTOS ADVERSOS: Experiencia de estudios clínicos en pacientes tratados con TEMODAL® cápsulas.
Pacientes con Glioblastoma Multiforme recién diagnosticado: La Tabla 1 presenta los eventos adversos (causalidad no determinada durante los estudios clínicos) ocurridos durante el tratamiento en pacientes con glioblastoma multiforme recién diagnosticado durante las fases de concomitancia y coadyuvante del tratamiento.
|
Tabla 1. TEMODAL® (TMZ) y radioterapia: Eventos adversos ocurridos durante el tratamiento concomitante y coadyuvante. Muy común (>1/10); Común (>1/100,<1/10); Poco común (>1/1000, <1/100) CIOMS III |
||
|
Sistema corporal |
TMZ+Radioterapia concomitante (n=288*) |
Terapia Coadyuvante con TMZ (n=224) |
|
Infecciones e Infestaciones |
||
|
Comunes: |
Candidiasis oral, herpes simple, infección, faringitis, infección de la herida |
Candidiasis oral, infección |
|
Poco Comunes: |
Herpes simple, herpes zoster, síntomas tipo influenza |
|
|
Trastornos del sistema sanguíneo y linfático |
||
|
Comunes: |
Leucopenia, linfopenia, neutropenia, trombocitopenia |
Anemia, neutropenia febril, leucopenia, trombocitopenia |
|
Poco comunes: |
Anemia, neutropenia febril |
Linfopenia, petequias |
|
Trastornos endocrinos |
||
|
Poco comunes: |
Cushingoide |
Cushingoide |
|
Trastornos metabólicos y nutricionales |
||
|
Muy comunes: |
Anorexia |
Anorexia |
|
Comunes: |
Hiperglicemia, disminución de peso |
Disminución de peso |
|
Poco comunes: |
Hipocalemia, elevación de la fosfatasa alcalina, aumento de peso |
Hiperglicemia, aumento de peso |
|
Trastornos psiquiátricos |
||
|
Comunes: |
Ansiedad, labilidad emocional, insomnio |
Ansiedad, depresión, labilidad emocional, insomnio |
|
Poco comunes: |
Agitación, apatía, trastorno de la conducta, depresión, alucinaciones |
Alucinaciones, amnesia |
|
Trastornos del sistema nervioso |
||
|
Muy comunes: |
Cefalea |
Cefalea, convulsiones |
|
Comunes: |
Mareo, afasia, limitación del equilibrio, alteración de la capacidad de concentración, confusión, disminución de la conciencia, convulsiones, daño en la memoria, neuropatía, parestesia, somnolencia, trastornos del habla, temblor |
Mareo, afasia, limitación del equilibrio, limitación de la capacidad de concentración, confusión, disfasia, hemiparesia, daño en la memoria, trastorno neurológico (NOS), neuropatía, neuropatía periférica, parestesia, somnolencia, trastornos del habla, temblor |
|
Poco comunes |
Ataxia, daño cognitivo, disfasia, trastorno extrapiramidal, marcha anormal, hemiparesis, hiperestesia, hipoestesia, trastorno neurológico (NOS), neuropatía periférica, estatus epiléptico. |
Ataxia, coordinación anormal, marcha anormal, hemiplejía, hiperestesia, trastornos sensitivos |
|
Trastornos oculares |
||
|
Comunes: |
Visión borrosa |
Visión borrosa, diplopía, defectos del campo visual |
|
Poco comunes: |
Dolor ocular, hemianopsia, trastornos visuales, disminución de la agudeza visual, defectos del campo visual |
Dolor ocular, ojo seco, disminución de la agudeza visual |
|
Trastornos del oído y laberinto |
||
|
Comunes: |
Daño auditivo |
Daño auditivo, tinnitus |
|
Poco comunes: |
Otalgia, hiperacusia, tinnitus, otitis media |
Sordera, otalgia, vértigo |
|
Trastornos cardiacos |
||
|
Poco comunes: |
Palpitaciones |
|
|
Trastornos vasculares |
||
|
Comunes: |
Edema, edema de miembros inferiores, hemorragia |
Edema de miembros inferiores, hemorragia, trombosis venosa profunda |
|
Poco comunes: |
Hipertensión, hemorragia cerebral |
Edema, edema periférico, embolismo pulmonar |
|
Trastornos respiratorios, torácicos y del mediastino |
||
|
Comunes: |
Tos, disnea |
Tos, disnea |
|
Poco comunes: |
Neumonía, infección respiratoria alta, congestión nasal |
Neumonía, sinusitis, infección respiratoria alta, bronquitis |
|
Trastornos gastrointestinales |
||
|
Muy comunes: |
Estreñimiento, náusea y vómito |
Estreñimiento, náusea y vómito |
|
Comunes: |
Dolor abdominal, diarrea, dispepsia, disfagia, estomatitis |
Diarrea, dispepsia, disfagia, boca seca, estomatitis |
|
Poco comunes: |
Distensión abdominal, incontinencia fecal, trastorno gastrointestinal (NOS), gastroenteritis, hemorroides |
|
|
Trastornos de la piel y del tejido subcutáneo |
||
|
Muy comunes: |
Alopecia, rash |
Alopecia, rash |
|
Comunes: |
Dermatitis, piel seca, eritema, prurito |
Piel seca, prurito |
|
Poco comunes: |
Reacción de fotosensibilidad, pigmentación anormal, exfoliación de la piel |
Eritema, pigmentación anormal, aumento de la sudoración |
|
Trastornos del tejido musculoesquelético y tejido conectivo |
||
|
Comunes: |
Artralgia, debilidad muscular |
Artralgia, dolor musculoesquelético, mialgia, debilidad muscular |
|
Poco comunes: |
Dolor de espalda, dolor musculoesquelético, mialgia, miopatía |
Dolor de espalda, miopatía |
|
Trastornos renales y urinarios |
||
|
Comunes: |
Aumento de la frecuencia urinaria, incontinencia urinaria |
Incontinencia urinaria |
|
Poco comunes: |
Disuria |
|
|
Trastornos del sistema reproductivo y de la glándula mamaria |
||
|
Poco comunes: |
Impotencia |
Amenorrea, dolor en senos, menorragia, hemorragia vaginal, vaginitis |
|
Trastornos generales y trastornos del sitio de la administración |
||
|
Muy comunes: |
Fatiga |
Fatiga |
|
Comunes: |
Fiebre, dolor, reacción alérgica, lesión por radiación, edema facial, alteración del gusto |
Fiebre, dolor, reacción alérgica, lesión por radiación, alteración del gusto |
|
Poco comunes: |
Enrojecimiento, oleadas de calor, astenia, estado agravado, rigor, decoloración de la lengua, parosmia, sed |
Astenia, estado agravado, dolor, rigor, alteraciones dentales, edema facial, alteración del gusto |
|
Investigación |
||
|
Comunes: |
Elevación de la SGPT |
Elevación de la SGPT |
|
Poco comunes: |
Elevación de: Gama glutamiltransferasa, enzimas hepáticas y SGOT |
|
|
*Un paciente aleatorizado al brazo de radioterapia sola recibió TEMODAL®+RT |
||
Resultados de Exámenes de Laboratorio: Se observó mielosupresión (neutropenia y trombocitopenia), los cuales son toxicidades limitantes conocidas de la dosis para la mayoría de los agentes citotóxicos, incluyendo TEMODAL®. Al combinar las anomalías en resultados de paraclínicos y eventos adversos en las fases concomitante y coadyuvante, se observaron anomalías Grado 3 o 4 en neutrófilos, incluyendo eventos neutropénicos, en
8% de los pacientes. Anomalías en plaquetas Grado 3 o 4, incluyendo eventos trombocitopénicos, fueron observadas en 14% de pacientes que recibieron TEMODAL®.
Pacientes adultos con Glioma recurrente o progresivo, o Melanoma maligno: En estudios clínicos, los efectos indeseables que ocurrieron con mayor frecuencia fueron los trastornos gastrointestinales, específicamente náuseas (43%) y vómito (36%). Estos efectos fueron usualmente Grado 1 o 2 CTC (de severidad leve a moderada) y fueron autolimitados o rápidamente controlados con terapia antiemética estándar. La incidencia de náusea severa y vómito fue del 4%. Mielosupresión severa, predominantemente trombocitopenia fue la reacción adversa limitante de la dosis y ocurrió en el 9% de todos los pacientes. Fueron reportadas anemia, neutropenia, leucopenia, linfopenia y pancitopenia. La mielosupresión fue usualmente predecible y ocurrió más a menudo en los primeros ciclos, con plaquetas y neutrófilos nadir ocurriendo de aparición tardía en el ciclo (usualmente durante los días 21 y 28) y la recuperación fue rápida (usualmente dentro de 1 a 2 semanas). No se observó evidencia de mielosupresión acumulativa.
Otros efectos adversos reportados frecuentemente incluyeron fatiga (22%), estreñimiento (17%) y cefalea (14%). Fueron reportadas también, anorexia (11%), diarrea (8%), rash, fiebre, astenia y somnolencia (6% cada una). Efectos colaterales menos comunes (2% a 5%) y en orden descendente de frecuencia fueron, dolor abdominal, dolor, mareo, pérdida de peso, malestar general, dispepsia, alopecia, rigor, prurito, disnea, alteración del gusto, parestesias y petequias.
Farmacocinética para Temozolomida oral: En un análisis de farmacocinética de la población en un ensayo clínico hubo 101 mujeres y 169 hombres para quienes el conteo nadir de neutrófilos estuvo disponible y 110 mujeres y 174 hombres para quienes el conteo nadir de plaquetas estuvo disponible. Hubo tasas más altas de neutropenia grado 4 (CAN < 500 células/µL), 12% versus 5%, y trombocitopenia (<20.000 células/ µL), 9% versus 3% en mujeres versus hombres en el primer ciclo de terapia. En una serie de 400 sujetos con glioma recurrente, la neutropenia grado 4 ocurrió en el 8% de las mujeres versus el 4% de los hombres y la trombocitopenia grado 4 ocurrió en el 8% de las mujeres versus el 3% de los hombres en el primer ciclo de terapia. En un estudio de 288 sujetos con glioblastoma multiforme recién diagnosticado, la neutropenia grado 4 ocurrió en el 3% de las mujeres versus el 0% de los hombres y la trombocitopenia grado 4 en el 1% de las mujeres versus el 0% de los hombres en el primer ciclo de terapia.
Pacientes tratados con TEMODAL® Polvo para inyección /Polvo para solución para infusión: TEMODAL® polvo para solución para infusión entrega una dosis de Temozolomida equivalente y exposición a ambos Temozolomida y MTIC como la concentración de TEMODAL® cápsulas correspondiente. Los eventos adversos probablemente relacionados al tratamiento que fueron reportados de los 2 estudios con la formulación IV (n=35) que no fueron reportados utilizando TEMODAL® cápsulas fueron aquellos relacionados con el sitio de infusión: dolor, irritación, prurito, calor, hinchazón y eritema en el sitio de infusión, así como hematoma.
Experiencia Post-Mercadeo con TEMODAL®: Durante la comercialización de TEMODAL®, se han reportado en muy raras ocasiones, casos de eritema multiforme, necrólisis epidérmica tóxica, síndrome de Steven-Johnson y reacciones alérgicas, incluyendo anafilaxia. Han sido reportados casos de hepatotoxicidad que incluyen elevaciones de enzimas hepáticas, hiperbilirrubinemia, colestasis y hepatitis. Daño hepático incluyendo falla hepática fatal, ha sido reportado muy rara vez (ver Sección Precauciones y advertencias).
También se han reportado en raras ocasiones infecciones oportunistas, incluyendo Neumonía por Pneumocystis carinii (PCP, por sus siglas en inglés); y ambos, casos de aparición y de reactivación de infecciones por citomegalovirus (CMV, por sus siglas en inglés). Se han reportado también, casos de reactivación de infecciones de hepatitis B, incluyendo algunos casos con desenlaces fatales (ver Sección Precauciones y advertencias). Casos de pneumonitis intersticial/pneumonitis y fibrosis pulmonar han sido reportados muy rara vez. En muy raras ocasiones se han observado casos de síndrome mielodisplásico y malignidades secundarias, incluyendo leucemia mieloide. Ha sido reportada pancitopenia prolongada que puede resultar en anemia aplasica, y en algunos casos ha dado lugar a un desenlace fatal. También ha sido reportada Diabetes insípida.
INTERACCIONES FARMACOLÓGICAS CON TEMODAL® ORAL (ver también PRECAUCIONES): En un estudio aleatorizado, abierto, de dos brazos, cruzado, pacientes con cáncer recibieron TEMODAL® 150 mg/m2/día con y sin tratamiento concomitante con ranitidina. No se observaron cambios en la farmacocinética o un incremento en el pH gástrico con TEMODAL® o MTIC debido a la ranitidina. La administración de TEMODAL® con alimentos resultó en una disminución estadísticamente significativa del 33% en el Cmax, un retraso de Tmax y una disminución pequeña pero estadísticamente significativa del 9% en el ABC (0 a 24). Dado que la actividad de TEMODAL® está relacionada con el ABC, más que con las concentraciones pico de TEMODAL®, el efecto de los alimentos no parece ser de importancia clínica. La coadministración de dexametasona, proclorperazina, fenitoina, carbamazepina, ondansetron, antagonistas de los receptores H2, o fenobarbital no alteraron la depuración de TEMODAL®. La coadministración con ácido valproico está asociada con una disminución pequeña pero estadísticamente significativa de la depuración de TEMODAL®.
El uso de TEMODAL® en combinación con otros agentes mielosupresores quimioterapéuticos puede incrementar la probabilidad de mielosupresión.
INTERFERENCIA CON PRUEBAS DE LABORATORIO: No hay datos disponibles.
PRECAUCIONES Y ADVERTENCIAS: Manejo por especialista. Chequeo hematológico periódico.
• Precauciones y advertencias:
Neumonía por Pneumocystis carinii: Se observó que los pacientes que recibieron TEMODAL® concomitantemente con radioterapia en un ensayo piloto en el esquema prolongado de 42 días, mostraron estar en un riesgo particular para desarrollar neumonía por Pneumocystis carinii (PCP, por sus siglas en inglés). Por ende, profilaxis contra neumonía por Pneumocystis carinii es requerida para todos los pacientes que reciban TEMODAL® concomitantemente con radioterapia durante el régimen de 42 días (con un máximo de 49 días) con independencia del recuento de linfocitos. Si se produce linfopenia, deben continuar con la profilaxis hasta que la recuperación de la linfopenia sea de un grado ≤ 1. Puede haber una mayor ocurrencia de PCP cuando se administra Temozolomida durante un régimen de dosificación más largo. Sin embargo, todos los pacientes que estén recibiendo Temozolomida particularmente los pacientes que reciben esteroides deberían ser monitoreados de cerca para el desarrollo de PCP relacionada con el régimen.
Terapia antiemética:
La náusea y vómito se asocian muy comúnmente con TEMODAL® y se proporciona la siguiente guía:
Pacientes con glioblastoma multiforme recién diagnosticado:
— Se recomienda profilaxis antiemética previa a la dosis inicial de Temozolomida concomitante,
— Es altamente recomendado profilaxis antiemética durante la fase de coadyuvancia.
Pacientes con gliomas recurrentes o progresivos: Los pacientes que hayan experimentado vómito severo (grado 3 o 4) en los ciclos previos pueden requerir terapia antiemética.
Parámetros de laboratorio para modificación de la dosis en Glioma maligno recurrente o progresivo o en melanoma maligno: Los pacientes tratados con TEMODAL® pueden sufrir mielosupresión, incluyendo pancitopenia prolongada, que puede resultar en anemia aplásica, que en algunos casos ha dado lugar a un desenlace fatal. En algunos casos, la exposición a medicamentos concomitantes asociados con la anemia aplásica, incluyendo carbamazepina, fenitoína y sulfametoxazol/trimetoprim, complica la evaluación. Antes de la administración de TEMODAL®, se debe cumplir con los siguientes parámetros de laboratorio: Recuento absoluto de neutrófilos (ANC, por sus siglas en inglés) 1,5 x 109/L y plaquetas 100 x 109/L. Debería realizarse un cuadro hemático completo en el día 22 (21 días después de la primera dosis) o dentro de las 48 horas siguientes a ese día, y semanalmente hasta que el ANC se encuentre sobre 1,5 x 109/L y el recuento de plaquetas exceda los 100 x 109/L. Sí el ANC cae a <1,0 x 109/L, o el número de plaquetas es <50 x 109/L durante cualquier ciclo, el siguiente ciclo debería reducirse a 50 mg/ m2. La dosis más baja recomendada es de 100 mg/m2 (ver sección Dosis y Administración para información completa sobre dosificación para glioma maligno recurrente o progresivo, melanoma maligno y glioblastoma multiforme recientemente diagnosticado).
Efecto de la función renal: La función renal determinada por la depuración estimada de creatinina no afecto la depuración de TEMODAL®.
Efecto de la función hepática: No fue observada una tendencia cuando la depuración de TEMODAL® fue trazada contra los parámetros individuales de la función hepática. Esto incluyó albumina sérica y proteína total así como también índices de enfermedad hepatocelular tales como fosfatasa alcalina, SGOT, SGPT y bilirrubina. La farmacocinética de Temozolomida en pacientes con enfermedad hepática leve a moderada (Child Pugh Clase I-II) fue similar a la observada en pacientes sin enfermedad hepática. La farmacocinética no está bien definida en pacientes con función hepática afectada severamente. Basados en la farmacocinética de la Temozolomida, las reducciones en la dosis no son requeridas en pacientes con falla hepática leve a moderada.
Daño hepático, incluyendo falla hepática fatal, ha sido reportado en pacientes tratados con temozolomida. Las pruebas de función hepática basales deberían ser realizadas previamente al inicio del tratamiento. Sí los resultados son anormales, el médico debería evaluar el beneficio/riesgo antes de iniciar temozolomida incluyendo el potencial de la insuficiencia hepática fatal. Para los pacientes en un ciclo de tratamiento de 42 días, las pruebas de función hepática se deberían repetir a mitad del tratamiento durante este ciclo. Para todos los pacientes, deberían revisarse las pruebas de función hepática después de cada ciclo de tratamiento. Para los pacientes con alteraciones significativas de la función hepática, los médicos deberían evaluar el balance beneficio/riesgo de continuar con el tratamiento. La toxicidad hepática puede aparecer varias semanas o más, después del último tratamiento con temozolomida.
Adicionalmente, se ha reportado hepatitis debido a reactivación del virus de hepatitis B (VHB), en algunos casos resultando en muerte. Los pacientes deberían ser evaluados para determinar infección VHB antes de la iniciación del tratamiento. Los pacientes con evidencia de infección VHB previa, deberían ser monitoreados para signos clínicos y de laboratorio de hepatitis o reactivación del virus VHB durante y por varios meses después del tratamiento con TEMODAL®. La terapia debería ser descontinuada para pacientes con evidencia de infección activa de hepatitis B.
Uso Pediátrico: No existe experiencia clínica con el uso de TEMODAL® en niños menores de 3 años de edad.
Uso en pacientes adultos mayores: En estudios clínicos, pacientes (>70 años de edad), parecen tener un incremento en el riesgo de neutropenia y trombocitopenia, en comparación con los pacientes más jóvenes.
Uso durante el embarazo y lactancia: En estudios preclínicos en ratas y en conejas, se demostró teratogenicidad y/o toxicidad fetal, cuando se administraron dosis de 150 mg/m2. No hay estudios en mujeres embarazadas. TEMODAL® puede ser utilizado durante el embarazo solo sí el beneficio potencial justifica el riesgo potencial para el feto. Las mujeres potencialmente fértiles, deberían ser advertidas para evitar el embarazo mientras están recibiendo TEMODAL®. Sí el uso de este medicamento durante el embarazo debe ser considerado o sí la paciente se embaraza mientras está tomando TEMODAL®, la paciente debería ser informada del riesgo potencial sobre el feto. No se sabe si TEMODAL® es excretado en la leche materna, por lo tanto, una decisión debería tomarse acerca de la descontinuación de la lactancia o de la administración de TEMODAL®.
Los efectos en ensayos en ambos, ratas y perros sugieren una fuerte posibilidad de efectos reproductivos adicionales, incluyendo infertilidad y posibles efectos en la descendencia resultando en daño genético a células germinales (puede ser posible una mutación en las células germinales que podría ser transmitida a la progenie). Considerando que estudios en ciclos múltiples indicaron toxicidad testicular, debería ser utilizada anticoncepción efectiva pacientes mujeres y hombres que estén recibiendo TEMODAL®. Considerando que Temozolomida es rápidamente convertida a MTIC (5-(3-metiltriacen-1-il) imidazol-4-carboxamida) su potencial tumorigénico no es inesperado. Esto es consistente con lo observado en otros agentes alquilantes incluyendo aquellos que producen MTIC. El potencial oncogénico total de Temozolomida en ratas parece ser especie-específico y no es significativamente diferente de otros medicamentos citotóxicos. Las cápsulas de TEMODAL® no deben ser abiertas o masticadas, deberían ser deglutidas completas con un vaso de agua. Sí la cápsula se daña evite el contacto del polvo con la piel o membranas mucosas. En caso de contacto con el polvo las manos deben lavarse.
Mantenga este medicamento fuera del alcance de los niños.
DOSIS Y ADMINISTRACIÓN: La dosificación de adultos y pediátricos se basa en los resultados de los Estudios Fase I C93-169, I93-114, C94-022 y I93-125 y el estudio fase III P00458.
• Pacientes adultos con Glioblastoma Multiforme recién diagnosticado:
Fase Concomitante: TEMODAL® se administra en dosis de 75 mg/m2 diario, durante 42 días concomitantemente con radioterapia (60 Gy administrados en 30 fracciones), seguido de TEMODAL® coadyuvante durante 6 ciclos. No se recomiendan reducciones de dosis; sin embargo, pueden ocurrir interrupciones de dosis según la tolerabilidad del paciente. La dosis de TEMODAL® puede ser continuada durante el período de concomitancia de 42 días hasta 49 días si se cumplen los siguientes criterios: Recuento absoluto de neutrófilos ≥1,5 x 109/L, número de plaquetas ≥ 100 x 109/L, toxicidad no hematológica según los criterios de toxicidad común (Common Toxicity Criteria; CTC) ≤ Grado 1 (exceptuando alopecia, náusea y vómito). Durante el tratamiento se debería obtener semanalmente un cuadro hemático completo. La administración de TEMODAL® debería ser interrumpida o suspendida en la fase de concomitancia de acuerdo a los criterios de toxicidad hematológica y no hematológica mencionados en la Tabla 2.
|
Tabla 2. Criterios de Interrupción o suspensión de la administración de TEMODAL® durante la fase de concomitancia con radioterapia |
||
|
Toxicidad |
TMZ Interrupcióna |
TMZ Suspensión definitiva |
|
Recuento absoluto de neutrófilos |
≥ 0,5 y <1,5 x 109/L |
< 0,5 x 109/L |
|
Recuento de plaquetas |
≥ 10 y <100 x 109/L |
< 10 x 109/L |
|
Toxicidad no hematológica (CTC) (exceptuando alopecia, náusea y vómito). |
CTC Grado 2 |
CTC Grado 3 o 4 |
|
a: El tratamiento concomitante con TMZ puede ser continuado cuando se cumplan todos los criterios enunciados a continuación: Recuento absoluto de neutrófilos ≥1,5 x 109/L, recuento de plaquetas ≥ 100 x 109/L, toxicidad no hematológica según los criterios de toxicidad común (CTC) ≤ Grado 1 (exceptuando alopecia, náusea y vómito). TMZ = TEMODAL®; CTC= Common Toxicity Criteria |
||
Fase Coadyuvante: Cuatro semanas luego de completar la fase de TEMODAL® + Radioterapia, TEMODAL® es administrado por 6 ciclos adicionales de tratamiento coadyuvante. La dosis en el ciclo 1 (Coadyuvante) es de 150 mg/m2 una vez al día durante 5 días, seguido de 23 días sin tratamiento. Al inicio del ciclo 2, se debe aumentar la dosis a 200 mg/m2 si la toxicidad no hematológica CTC durante el ciclo 1 fue grado ≤ 2 (excepto por alopecia, nausea y vómito), recuento absoluto de neutrófilos (ANC) ≥1,5 x 109/L y recuento de plaquetas ≥ 100 x 109/L. Sí la dosis no es aumentada en el ciclo 2, no debería realizarse en los ciclos subsecuentes. La dosis permanecerá en 200 mg/m2 por día para los primeros 5 días de cada ciclo subsecuente excepto si se presenta toxicidad. Las reducciones de dosis durante la fase de coadyuvancia deberían realizarse de acuerdo a las Tablas 3 y 4.
Durante el tratamiento debería realizarse un cuadro hemático completo en el día 22 (21 días después de la primera dosis de TEMODAL®). La dosis de TEMODAL® debería ser reducida o suspendida de acuerdo a la Tabla 4.
|
Tabla 3. Niveles de dosis de TEMODAL® para tratamiento coadyuvante |
||
|
Niveles de Dosis |
Dosis mg/m2/día |
Comentarios |
|
-1 |
100 |
Reducción para toxicidad previa |
|
0 |
150 |
Dosis durante el ciclo 1 |
|
1 |
200 |
Dosis durante los ciclos 2 a 6 en ausencia de toxicidad |
|
Tabla 4. Reducción o suspensión de la dosis de TEMODAL® durante el tratamiento coadyuvante |
||
|
Toxicidad |
Reducir TMZ 1 nivel de dosis a |
Suspender TMZ |
|
Recuento absoluto de neutrófilos |
< 1,0 x 109/L |
b |
|
Recuento de plaquetas |
< 50 x 109/L |
b |
|
Toxicidad no hematológica CTC (excepto para alopecia, nausea, vómito) |
CTC Grado 3 |
CTC Grado 4b |
|
a: Niveles de dosis de TMZ listados en la Tabla 3 b: TMZ debe ser suspendido si se requiere una reducción de dosis a < 100 mg/m2 o si recurre el mismo grado 3 de toxicidad no hematológica (excepto por alopecia, nausea, vómito) después de la reducción de dosis. TMZ = TEMODAL®; CTC= Common Toxicity Criteria |
||
Adultos con Glioma recurrente o progresivo o melanoma maligno: En pacientes que no hayan sido tratados previamente con quimioterapia, TEMODAL® debería administrarse a una dosis de 200 mg/m2 una vez al día, por 5 días, en ciclos cada 28 días. En pacientes previamente tratados con quimioterapia, la dosis inicial es de 150 mg/m2 una vez al día, e incrementada en el segundo ciclo a 200 mg/m2 diariamente, siempre y cuando el número absoluto de neutrófilos (ANC) sea ≥ 1,5 x 109/L y el número de plaquetas sea ≥ 100 x 109/L en el día 1 del siguiente ciclo. La modificación de la dosis para TEMODAL® debería estar basada en la toxicidad de acuerdo a los conteos nadir de ANC o plaquetas.
Pacientes Pediátricos con Glioma recurrente o progresivo: En pacientes de 3 años de edad o mayores, TEMODAL® se recomienda, en una dosis de 200 mg/m2 una vez al día, durante 5 días, en ciclos cada 28 días. Los pacientes pediátricos previamente tratados con quimioterapia deberían recibir una dosis inicial de 150 mg/m2 una vez al día, por 5 días, con un incremento de la dosis a 200 mg/m2 una vez al día por 5 días en el siguiente ciclo, si no hay toxicidad hematológica.
TEMODAL® cápsulas puede ser administrado fuera del horario de las comidas, sin embargo, la administración una hora antes de una comida puede ayudar a reducir las náuseas. La terapia antiemética puede ser administrada antes o después de la administración de TEMODAL®.
El tratamiento con TEMODAL® puede continuarse hasta la progresión de la enfermedad por un máximo de 2 años.
TEMODAL® polvo para inyección/polvo para solución para infusión: Cada vial de TEMODAL® polvo para inyección/polvo para solución para infusión contiene polvo liofilizado de Temozolomida. Cuando es reconstituido con 41 mL de agua estéril para inyección, la solución resultante contiene 2,5 mg/mL de Temozolomida. Los viales deberían ser girados sutilmente pero no agitados. Los viales deberían ser inspeccionados y cualquier vial que contenga material particulado visible no debería ser utilizado. El producto reconstituido debe ser utilizado dentro de 14 horas incluyendo el tiempo de infusión.
Utilizando una técnica aséptica, tome 40 mL de cada vial para completar la dosis total y transfiéralo a una bolsa de infusión vacía de 250 mL. TEMODAL® polvo para inyección/polvo para solución para infusión debería ser administrado por infusión utilizando una bomba por un periodo de 90 minutos. TEMODAL® polvo para inyección/polvo para solución para infusión debería ser administrado solo por infusión intravenosa.
TEMODAL® polvo para inyección/polvo para solución para infusión puede ser administrado en la misma línea IV con una inyección de cloruro de sodio 0,9%. Es incompatible con soluciones de dextrosa.
Dado que no hay datos disponibles sobre la compatibilidad de TEMODAL® polvo para inyección/polvo para solución para infusión con otras sustancias o aditivos intravenosos, no deberían administrarse de forma intravenosa otros medicamentos simultáneamente a través de la misma línea intravenosa.
SOBREDOSIS: Se han evaluado clínicamente, en pacientes, dosis de 500, 750, 1.000 y 1.250 mg/m2 (dosis total por ciclo). La toxicidad limitante de la dosis fue hematológica y se reportó con cualquier dosis, pero se espera que sea más severa con dosis altas. Un paciente tomó una sobredosis de 10.000 mg (dosis total en un único ciclo de 5 días) y los eventos reportados fueron pancitopenia, pirexia, falla multiorgánica y muerte. Existen reportes de pacientes que han tomado tratamientos mayores a 5 días (hasta 64 días) con la aparición de eventos adversos que incluyen supresión de la médula ósea, la cual en algunos casos fue severa y prolongada, e infecciones y llevaron a la muerte. En el evento de una sobredosis, se requiere evaluación hematológica. Se deberían proveer medidas de soporte según sea necesario.
INFORMACIÓN PRECLÍNICA: Temozolomida sufre rápidamente conversión no enzimática a pH fisiológico para el compuesto activo, MTIC. La citotoxicidad de MTIC se cree que se debe fundamentalmente a la alquilación en la posición O6 de la guanina con una alquilación adicional que se produce en la posición N7. Las lesiones citotóxicas que se desarrollan posteriormente se cree que conllevan una reparación aberrante del aducto metilo.
Los estudios de toxicidad de dosis única de temozolomida fueron conducidos en ratones, ratas y perros. Las dosis LD50 estimadas para la vía oral fueron moderadamente más altas en las ratas (aproximadamente 1900 mg/m2) que en los ratones (aproximadamente 1000 mg/m2). La dosis mínima letal en perros fue 600 mg/m2. En los estudios de dosis única, los signos clínicos de toxicidad y muerte se presentaron generalmente con retraso, reflejando una toxicidad retardada a los tejidos que normalmente proliferan más rápidamente resultando en deterioro general de una función de un órgano; la toxicidad es consistente con lo que se espera de un agente alquilante.
Un solo ciclo (administración durante 5 días, 23 días sin tratamiento) y de tres y seis estudios de toxicidad de ciclo se llevaron a cabo en ratas y perros. Las concentraciones plasmáticas de temozolomida estuvieron relacionadas con la dosis e independientes del género, sin evidencia de acumulación. Los estudios de tres y seis ciclos revelaron toxicidad similar a la observada en los estudios de ciclo único, con la excepción de cambios neoplásicos en ratas en los estudios de tres y seis ciclos. Todos los hallazgos eran típicos de los que se esperan de un agente alquilante. En consecuencia, los estudios de toxicidad se limitaron a 6 ciclos. Temozolomida se absorbe rápidamente tras la administración oral a ratas y perros y la radiactividad derivada del fármaco es eliminada rápidamente en la orina. La exposición sistémica a la dosis terapéutica en seres humanos es similar a la de la rata y el perro. Temozolomida demostró en experimentos con animales que cruza fácilmente la barrera hematoencefálica. La temozolomida penetró rápidamente en el cerebro de ratas después de la administración oral, alcanzando el Tmáx en 1 hora. El grado de penetración de la temozolomida en el cerebro de rata osciló entre 35 y 39% basado en la proporción ABC cerebro a plasma. Se encontraron resultados similares para la penetración de la temozolomida en el líquido cefalorraquídeo (LCR) de perros. Las concentraciones cerebrales y plasmáticas de temozolomida disminuyeron en paralelo.
No se han realizado estudios de carcinogenicidad de la temozolomida. Sin embargo, los resultados del estudio de seis ciclos en ratas se pueden utilizar para evaluar el potencial carcinogénico de la temozolomida. Las neoplasias observadas en el estudio de seis ciclos en rata incluyen, carcinoma de mama, queratoacantoma de la piel, adenoma de células basales y una variedad de neoplasias mesenquimales. No se observaron tumores o alteraciones preneoplásicas en los estudios en perros.
Teniendo en cuenta que la temozolomida se convierte fácilmente en MTIC, su potencial tumorigénico no es inesperado. Esto es consistente con lo que se ha observado con otros agentes alquilantes, incluyendo los que producen MTIC. El potencial oncogénico general de temozolomida en ratas parece ser específico de la especie y no significativamente diferente de otros fármacos anticancerígenos alquilantes comercializados.
Resultados de las pruebas de Ames/Salmonella y HPBL mostraron una respuesta positiva de mutagenicidad. Temozolomida causó aumentos relacionados con la dosis en la frecuencia de mutación en 4 de 5 cepas de Salmonella typhimurium, con y sin activación metabólica. La frecuencia de mutación a la mayor concentración de exposición varió desde 2,3 hasta 14,6 veces por encima de los controles. Temozolomida también produjo aberraciones cromosómicas en un ensayo en linfocitos periféricos humanos con o sin activación metabólica utilizando enzimas de hígado de rata.
Se realizaron pruebas de toxicidad reproductiva en dos estudios de hallazgos de rangos de dosis en ratas y conejos seguidos por un estudio de toxicidad embrio-fetal en ratas. No se observó toxicidad materna significativa y las tasas de embarazo no fueron afectadas en estas especies. La dosificación no influenció las tasas de implantación o duración de la gestación. Los hallazgos de estos estudios indican que temozolomida, como otros agentes alquilantes, tiene el potencial para producir embrio letalidad y malformaciones en ratas y conejos.
Se encontró que Temozolomida era más tóxica en la rata y el perro que en los seres humanos, como el régimen de dosis terapéutica (200 mg/m2), que ha sido bien tolerado en los seres humanos, se aproxima a la dosis letal mínima después de múltiples dosis en ratas y perros. Las ratas parecen ser muy sensibles a los efectos tumorigénicos de temozolomida. Diferencias entre especies en la capacidad para reparar el daño del ADN proporciona razón plausible de las diferencias en la toxicidad observadas entre perros y seres humanos en relación con las ratas expuestas esencialmente a los mismos regímenes de dosificación.
En general, el perfil de toxicidad de temozolomida es coherente con su mecanismo de acción y no parece ser mayor que el de otros agentes alquilantes utilizados en el tratamiento del cáncer. Los objetivos principales de toxicidad son órganos que relativamente tienen células que proliferan rápidamente e incluyen la médula ósea, el sistema linforreticular, los testículos y el tracto gastrointestinal.
El perfil toxicológico preclínico de temozolomida para la administración intravenosa es comparable a la de la formulación oral (cápsula) y consistente con la de otros agentes anticancerígenos alquilantes comercializados.
Mientras que la formulación intravenosa produce irritación local en el sitio de la inyección en ambos conejos y ratas, la irritación fue transitoria y no estuvo asociada con el daño tisular duradero.
OTRA INFORMACIÓN RELEVANTE DE SEGURIDAD: No hay datos disponibles.
ABUSO Y DEPENDENCIA DE DROGAS: No Aplicable.
DESCRIPCIÓN DEL PRODUCTO: Temozolomida (SCH 52365), el ingrediente activo de TEMODAL® es un derivado imidazotetrazina. Su nombre químico es 3,4-dihidro-3-metil-4- oxoimidazo [5,1-d]-as-tetrazina-8-carboxamida. El peso molecular es 194.15, la fórmula molecular es C6H6N6O2 y la fórmula estructural es:
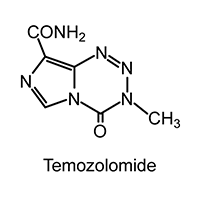
La molécula de temozolomida es estable a pH ácido, (<5) y por lo tanto puede ser administrada por vía oral. Temozolomida es rápidamente hidrolizada al activo 5-(3-metiltriazen-1-il) imidazol-4-carboxamida (MTIC) a valores de pH neutro, teniendo lugar la hidrólisis incluso más rápido a pH alcalino.
Cada CÁPSULA de TEMODAL® contiene 20 mg, 100 mg, 140 mg o 250 mg de Temozolomida. Cada vial de TEMODAL® contiene 100 mg de POLVO liofilizado de Temozolomida.
PRESENTACIÓN: TEMODAL® (Temozolomida): Polvo liofilizado (Temozolomida 100 mg) en vial de vidrio (Reg. San. INVIMA 2010M-0010658). Caja por 5 sachets de 1 cápsula de 20 mg (Reg. San. INVIMA 2017M-014897-R2). Caja por 5 sachets de 1 cápsula de 100 mg (Reg. San. INVIMA 2017M-0000094- R2). Caja por 5 sachets de 1 cápsula de 140 mg (Reg. San. INVIMA 2009M-0009548). Caja por 5 sachets de 1 cápsula de 250 mg (Reg. San. INVIMA 2016M-014875-R2).
Fecha de revisión del documento: Este documento fue revisado por última vez en Marzo de 2015.
WPI-MK7365-MTL-032015
MERCK SHARP & DOHME
Bogotá, D.C. - Colombia
ALMACENAMIENTO: Las cápsulas de 20 y 100, 140 y 250 mg deben almacenarse a temperatura inferior a 30 °C en su envase y empaque original. El vial de TEMODAL® de 100 mg debe almacenarse a temperatura entre 2°C - 8°C en su envase original, después de reconstituido, usar la solución dentro de las 14 horas siguientes a 25°C, incluyendo el tiempo de infusión.


