

VOKANAMET
CANAGLIFLOZINA, METFORMINA
Tabletas recubiertas
Tabletas recubiertas, 150/850 mg/mg
Tabletas recubiertas, 150/1000 mg/mg
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
FORMAS FARMACÉUTICAS Y CONCENTRACIONES: Las tabletas de 150 mg de canagliflozina/850 mg de clorhidrato de metformina son tabletas de liberación inmediata, con forma de cápsula, recubiertos con una película, de color amarillo claro con la inscripción “CM” de un lado y “418” del otro lado.
Las tabletas de 150 mg de canagliflozina/1000 mg de clorhidrato de metformina son tabletas de liberación inmediata, con forma de cápsula, recubiertos con una película, de color púrpura con la inscripción “CM” de un lado y “611” del otro lado.
Consulte los excipientes en la sección Lista de excipientes.
INDICACIONES:
VOKANAMET™ está indicado en adultos de 18 años en adelante que padezcan de diabetes mellitus del tipo 2 como complemento de la dieta y del ejercicio para mejorar el control glucémico:
• En pacientes que no están controlados de manera adecuada en sus dosis máximas toleradas de metformina sola.
• En pacientes en sus dosis máximas toleradas de metformina junto con otros productos medicinales para reducir la glucosa, entre ellos la insulina, cuando estos no proporcionan un control adecuado de la glucemia (ver sección Advertencias y Precauciones, Interacciones y Propiedades farmacodinámicas para obtener información sobre los datos disponibles sobre diferentes terapias adicionales).
• En pacientes que ya reciben el tratamiento con la combinación de canagliflozina y metformina como tabletas separados.
PROPIEDADES FARMACOCINÉTICAS: La farmacocinética de canagliflozina es esencialmente similar en sujetos sanos y en pacientes con diabetes tipo 2. Después de la administración de una dosis oral única de 100 mg y 300 mg en sujetos sanos, la canagliflozina se absorbe rápidamente, con concentraciones plasmáticas máximas (mediana de Tmax) ocurriendo de 1 a 2 horas después de la dosis. La Cmax plasmática y la AUC de la canagliflozina aumentaron de una forma proporcional a la dosis desde 50 mg a 300 mg. La vida medica terminal aparente (t1/2) (expresada como promedio ± desviación estándar) fue de 10.6 horas ± 2.13 horas y 13.1 ± 3.28 horas para las dosis de 100 y 300 mg, respectivamente. El estado estacionario se alcanzó después de administrar durante 4 a 5 días una dosis al día de canagliflozina de 100 mg a 300 mg. La canagliflozina no exhibe una farmacocinética dependiente del tiempo y se acumula en el plasma hasta un 36% tras dosis múltiples de 100 mg y 300 mg.
Un estudio de bioequivalencia en sujetos sanos demostró que las tabletas combinadas de VOKANAMET™ 150 mg/850 mg y 150 mg/1000 mg son bioequivalentes a la co-administración de las dosis correspondientes de canagliflozina y de clorhidrato de metformina como tabletas individuales.
La administración de la combinación de dosis fijas de VOKANAMET™ 150 mg/1000 mg con los alimentos no resultó en cambios en la exposición total de canagliflozina. No hubo cambios en el AUC de metformina; sin embargo, la concentración plasmática máxima promedio de la metformina disminuyó en un 16% cuando se administró con los alimentos. Se observó un retraso para alcanzar la concentración plasmática máxima para ambos componentes (2 horas para canagliflozina y 1 hora para metformina) bajo condiciones de alimentación. No es probable que estos cambios sean clínicamente importantes. Como se recomienda que metformina se administre con los alimentos para reducir la incidencia de los efectos secundarios gastrointestinales, se recomienda que VOKANAMET™ se administre con los alimentos para reducir la intolerabilidad gastrointestinal asociada con metformina.
Absorción:
Canagliflozina: La biodisponibilidad oral absoluta promedio de canagliflozina es aproximadamente 65%. La co-administración de un alimento alto en grasas con canagliflozina no tuvo efecto sobre la farmacocinética de la canagliflozina; por lo tanto, la canagliflozina puede tomarse con o sin alimentos (ver sección Posología y Administración).
Metformina: Después de una dosis oral de metformina, el Tmax se alcanza en 2.5 horas. La biodisponibilidad absoluta de una tableta de 500 mg o 850 mg de metformina es aproximadamente del 50 - 60% en los sujetos sanos. Después de una dosis oral, la fracción no absorbida recuperada en las heces fue 20 - 30%.
Después de la administración oral, la absorción de la metformina es saturable e incompleta. Se asume que la farmacocinética de la absorción de metformina es no lineal. Con programas de dosificación y dosis de metformina usuales, las concentraciones plasmáticas del estado estacionario se alcanzaron dentro de las 24-48 horas y generalmente son inferiores a 1 µg/mL. En ensayos clínicos controlados, los niveles máximos plasmáticos de metformina (Cmax) no excedieron 5 µg/mL, incluso a dosis máximas.
Los alimentos disminuyen la extensión y retrasan levemente la absorción de metfomina. Después de la administración de una dosis de 850 mg, se observó una disminución del 40% de la concentración plasmática máxima, una disminución del 25% en el AUC y una prolongación de 35 minutos del tiempo para alcanzar la concentración plasmática máxima. Se desconoce la relevancia clínica de esta disminución.
Distribución:
Canagliflozina: El volumen de distribución promedio en el estado estacionario de la canagliflozina después de una única infusión intravenosa en pacientes sanos fue de 83.5 L, sugiriendo una extensa distribución tisular. La canagliflozina se une fuertemente a las proteínas plasmáticas (99%), principalmente a la albúmina. La unión a las proteínas es independiente de las concentraciones plasmáticas de la canagliflozina. La unión a las proteínas plasmáticas no se alteró significativamente en pacientes con insuficiencia renal o hepática.
Metformina: La unión a proteínas plasmáticas es insignificante. La metformina se divide en los eritrocitos. El pico sanguíneo es menor que el pico plasmático y aparece aproximadamente al mismo tiempo. Es más probable que los glóbulos rojos representen un compartimento secundario de distribución. El Vd promedio osciló entre 63 - 276 litros.
Metabolismo:
Canagliflozina: La O-glucuronidación es la vía principal de eliminación metabólica de la canagliflozina, el cual es glucuronizada principalmente por UGT1A9 y UGT2B4 hasta dos metabolitos O-glucuronidos inactivos. Se observaron incrementos en el AUC de canagliflozina (26% y 18%) en sujetos portadores el alelo UGT1A9*3 y el alelo UGT2B4*2, respectivamente. No se espera que estos incrementos de la exposición a la canagliflozina sean clínicamente importantes. En humanos, el metabolismo (oxidativo) de la canagliflozina mediado por CYP3A4 es mínimo (aproximadamente 7%).
Metformina: La metformina se excreta sin cambios en la orina. En humanos, no se han identificado metabolitos.
Eliminación:
Canagliflozina: Después de la administración de una única dosis oral de canagliflozina [14C] a sujetos sanos, se recuperó en las heces 41.5%, 7.0% y 3.2% de la dosis radiactiva administrada como canagliflozina, un metabolito hidroxilado, y un metabolito O-glucurónido, respectivamente. La circulación enterohepática de canagliflozina fue insignificante.
En orina, se excretó aproximadamente el 33% de la dosis radiactiva administrada, principalmente como metabolitos O-glucurónido (30.5%). Menos del 1% de la dosis se excretó por la orina como canagliflozina inalterada.
El aclaramiento renal de las dosis de 100 mg y 300 mg osciló de 1.30 a 1.55 mL/min.
La canagliflozina es un fármaco de aclaramiento bajo, con un aclaramiento sistémico promedio de aproximadamente 192 mL/min en pacientes sanos después de la administración intravenosa.
Metformina: El aclaramiento renal de metformina es > 400 mL/min, indicando que la metformina se elimina por filtración glomerular y secreción tubular. Después de una dosis oral, la vida media terminal aparente es aproximadamente 6.5 horas. Cuando la función renal está deteriorada, disminuye el aclaramiento renal en proporción al de la creatinina y así la vida media de eliminación se prolonga, conduciendo a niveles incrementados de metformina en el plasma.
Poblaciones especiales:
Insuficiencia renal: No se realizaron estudios que caractericen la farmacocinética de canagliflozina y metformina después de la administración de VOKANAMET™ en pacientes con insuficiencia renal. Como la metformina está contraindicada en pacientes con insuficiencia renal, la administración de VOKANAMET™ también está contraindicada en pacientes con insuficiencia renal (por ejemplo, creatinina sérica ≥ 1.5 mg/dL (varones) o ≥ 1.4 mg/dL (mujeres) o aclaramiento de creatinina anormal) (ver sección Contraindicaciones y Advertencias y Precauciones).
Canagliflozina: Un estudio abierto de dosis única evaluó la farmacocinética de canagliflozina 200 mg en sujetos con diversos grados de insuficiencia renal [clasificada utilizando la fórmula de Modificación de la Dieta en la Enfermedad Renal (MDRD)-TFGe] en comparación con sujetos sanos. El estudio incluyó 3 sujetos con función renal normal (TFGe ≥ 90 mL/min/1.73 m2), 10 sujetos con insuficiencia renal leve (TFGe 60 a < 90 mL/min/1.73 m2), 9 sujetos con insuficiencia renal moderada (TFGe 30 a < 60 mL/min/1.73 m2) y 10 pacientes con insuficiencia renal severa (TFGe 15 a < 30 mL/min/1.73 m2) así como 8 sujetos con enfermedad renal en estado terminal con hemodiálisis.
La Cmáx de la canagliflozina se incrementó moderadamente en un 13%, 29% y 29% en sujetos con insuficiencia renal leve, moderada y grave, respectivamente, pero no en los pacientes con hemodiálisis. En comparación con los pacientes sanos, el AUC plasmático de canagliflozina se incrementó en aproximadamente 17%, 63% y 50% en sujetos con insuficiencia renal leve, moderada y severa, respectivamente, pero fue similar en pacientes con con enfermedad renal en estado terminal y en sujetos sanos (ver sección Posología y Administración, Advertencias y Precauciones y Reacciones adversas).
La eliminación de canagliflozina mediante hemodiálisis es insignificante.
Metformina: En pacientes con función renal disminuida (basados en la medición del aclaramiento de la creatinina), la vida media de metformina en sangre y plasma se prolonga y el aclaramiento renal disminuye en proporción a la disminución del aclaramiento de la creatinina (ver sección Contraindicaciones y Advertencias y Precauciones).
Insuficiencia hepática: No se realizaron estudios que caractericen la farmacocinética de canagliflozina y metformina después de la administración de VOKANAMET™ en pacientes con insuficiencia hepática. Sin embargo, el uso solo de metformina en pacientes con insuficiencia hepática ha sido asociada con algunos casos de acidosis láctica. Por lo tanto, el uso de VOKANAMET™ no se recomienda en pacientes con insuficiencia hepática (ver sección Advertencias y Precauciones).
Canagliflozina: Respecto a los sujetos con función hepática normal, la proporción geométrica promedio para la Cmáx y el AUC∞ de canagliflozina fueron 107% y 110%, respectivamente, en sujetos con Child-Pugh de clase A (insuficiencia hepática leve) y 96% y 111%, respectivamente, en sujetos con Child-Pugh de clase B (insuficiencia hepática moderada) después de la administración de una dosis única de 300 mg de canagliflozina.
Estas diferencias no son consideras clínicamente significativas.
Ancianos (≥ 65 años de edad): No se realizaron estudios que caractericen la farmacocinética de canagliflozina y metformina después de la administración de VOKANAMET™ en pacientes geriátricos. El tratamiento con VOKANAMET™ no debe iniciarse en ancianos sin monitoreo inicial y de rutina de la función renal (ver sección Advertencias y Precauciones).
Canagliflozina: La edad no tuvo efecto clínicamente significativo sobre la farmacocinética de canagliflozina en base a un análisis farmacocinético poblacional (ver sección Posología y Administración, Advertencias y Precauciones y Reacciones Adversas).
Población pediátrica (< 18 años de edad): No se realizaron estudios que caractericen la farmacocinética de canagliflozina y metformina después de la administración de VOKANAMET™ en pacientes pediátricos.
Otras poblaciones: No es necesario ajustar la dosis en base al género, raza/etnia o índice de masa corporal. Estas características no tuvieron efecto clínicamente significativo sobre la farmacocinética de canagliflozina en base a un análisis farmacocinético poblacional.
PROPIEDADES FARMACODINÁMICAS:
Grupo farmacoterapéutico: Combinaciones de fármacos hipoglicemiantes orales, código ATC: A10BD16.
Mecanismo de acción: VOKANAMET™ combina dos agentes orales antihiperglucémicos con mecanismos de acción complementarios para mejorar el control glucémico en pacientes con diabetes tipo 2: La canagliflozina, un inhibidor del co-transportador de sodio-glucosa tipo 2 (SGLT2) y el clorhidrato de metformina, un miembro de la clase biguanida. La metformina disminuye la producción de glucosa hepática, disminuye la absorción intestinal de glucosa y mejora la sensibilidad a la insulina incrementando la captación y utilización de la glucosa periférica.
Canagliflozina: El co-transportador de sodio-glucosa 2 (SGLT2), expresado en los túbulos renales proximales, es responsable de la mayor parte de la reabsorción de la glucosa filtrada desde la luz tubular. Los pacientes con diabetes han demostrado tener reabsorción renal de glucosa elevada el cual puede contribuir a las concentraciones elevadas de glucosa persistente. La canagliflozina es un inhibidor de SGLT2 activo por vía oral. La canagliflozina, mediante la inhibición de SGLT2, reduce la reabsorción de la glucosa filtrada y reduce el umbral renal de la glucosa (RTG) y por lo tanto aumenta la excreción urinaria de glucosa (EUG), disminuyendo las concentraciones plasmáticas elevadas de glucosa mediante este mecanismo independiente de la insulina en pacientes con diabetes tipo 2. El aumento de la EUG con la inhibición de SGLT2 también se traduce en una diuresis osmótica, con el efecto diurético que conduce a una reducción de la presión sanguínea sistólica; el aumento de la EUG resulta en una pérdida de calorías y por lo tanto una reducción en el peso corporal, como se ha demostrado en estudios de pacientes con diabetes tipo 2.
La acción de la canagliflozina para aumentar la EUG directamente dismuyendo la glucosa plasmática es independiente de la insulina. En estudios clínicos con canagliflozina se han observado mejorías en la evaluación del modelo de homeostasis para la función de las células beta (células beta HOMA) y una respuesta mejorada de secreción de insulina de las células beta a un desafío de alimentos combinados.
Metformina: La metformina es una biguanida con efectos antihiperglucémicos, disminuyendo la glucosa plasmática basal y postprandial. No estimula la secreción de insulina y por lo tanto no produce hipoglucemia.
La metformina puede actuar mediante tres mecanismos:
• Mediante la reducción de la producción de glucosa hepática inhibiendo la gluconeogénesis y la glucogenólisis.
• En los músculos, al aumentar modestamente la sensibilidad a la insulina, mejorando la captación y utilización de la glucosa periférica.
• Mediante el retraso de la absorción intestinal de la glucosa.
La metformina estimula la síntesis del glucógeno intracelular actuando sobre la sintasa glucógena. La metformina aumenta la capacidad de transporte de los tipos específicos de transportadores de glucosa de membrana (GLUT-1 y GLUT-4). En estudios clínicos, se asoció a la metformina con un peso corporal estable o con una pérdida modesta de peso.
En los humanos, independientemente de su acción sobre la glucemia, la metformina tiene efectos favorables sobre el metabolismo de los lípidos. Esto ha sido demostrado con dosis terapéuticas en estudios clínicos controlados, de medio plazo o largo plazo: La metformina reduce el colesterol total, el LDL-C y los niveles de triglicéridos.
INFORMACIÓN FARMACÉUTICA:
Lista de excipientes: El núcleo de la tableta de canagliflozina contiene croscarmelosa de sodio, hipromelosa, estearato de magnesio (de origen vegetal) y celulosa microcristalina y agua purificada. Los tabletas se terminan con un recubrimiento con película disponible comercialmente que consta de los siguientes excipientes: Macrogol 3350, alcohol polivinílico (parcialmente hidrolizado), talco, dióxido de titanio, óxido de hierro amarillo (solamente las tabletas de 150 mg/850 mg), óxido de hierro rojo (solamente las tabletas de 150 mg/1000 mg) y óxido de hierro negro (solamente las tabletas de 150 mg/1000 mg).
Incompatibilidades: No procede.
Período de validez: Observe la fecha de caducidad en el embalaje exterior.
CONTRAINDICACIONES: VOKANAMET™ está contraindicado en pacientes con:
• Deterioro renal moderado y grave (pacientes con TFGe < 60 mL/min/1.73 m2 o CrCl < 60 mL/min)
• Acidosis metabólica aguda, que incluye cetoacidosis diabética (CAD), con o sin coma.
• Antecedentes de hipersensibilidad a la canagliflozina, a la metformina o a cualquiera de los excipientes.
EMBARAZO, LACTANCIA Y FERTILIDAD:
Embarazo: No existen estudios adecuados y bien controlados con VOKANAMET™ o sus componentes individuales en mujeres embarazadas.
Los estudios con canagliflozina en animales no indican efectos dañinos directos o indirectos respecto a la toxicidad reproductiva (ver sección Información No Clínica).
Una cantidad limitada de datos del uso de metformina en mujeres embarazadas no indica un incremento del riesgo de anormalidades congénitas. Los estudios con metformina en animales no indican efectos dañinos respecto al embarazo, el desarrollo embrionario o fetal, el parto o el desarrollo postnatal (ver sección Información No Clínica).
Durante el embarazo, considerar terapias alternativas adecuadas, especialmente durante el segundo y tercer trimestre. VOKANAMET™ debe usarse durante el embarazo, solamente si el beneficio potencial justifica el riesgo potencial para el feto.
Lactancia: No se realizaron estudios en animales que amamantan con los principios activos combinados de VOKANAMET™.
Los datos farmacodinámicos/toxicológicos en animales disponibles han demostrado excreción de canagliflozina en la leche. Se desconoce si canagliflozina se excreta en la leche humana. Los estudios en ratas que amamantan muestran que la metformina se excreta en la leche y alcanza niveles comparables con los del plasma. La metformina se excreta en la leche humana.
No puede excluirse un riesgo para el lactante. Se debe tomar la decisión de discontinuar la lactancia o discontinuar la terapia con VOKANAMET™, tomando en cuenta el beneficio de la lactancia para el niño y el beneficio de la terapia para la madre (ver sección Información No Clínica).
Fertilidad: No se ha estudiado el efecto de VOKANAMET™ sobre la fertilidad en humanos. En los estudios con animales no se observaron efectos de la canagliflozina sobre la fertilidad (ver sección Información No Clínica).
La metformina no afectó la fertilidad de las ratas machos y hembras cuando se les administró dosis tan altas como 600 mg/kg/día, lo cual es aproximadamente a 3 veces la dosis diaria máxima recomendada en humanos en base a comparaciones del área de superficie corporal.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: VOKANAMET™ no tiene influencia conocida sobre la capacidad para conducir y utilizar máquinas. Sin embargo, se debe alertar a los pacientes del riesgo de hipoglucemia cuando VOKANAMET™ es usado como complemento a la terapía con insulina o un secretagogo de insulina, y del riesgo elevado de reacciones adversas relacionadas al volumen intravascular reducido, como mareo postural (ver sección Posología y Administración, Advertencias y Precauciones y Reacciones Adversas).
EFECTOS FARMACODINÁMICOS: Después de dosis orales únicas y múltiples de canagliflozina en pacientes con diabetes tipo 2, se observaron disminuciones dependientes de la dosis del RTG e incrementos en la EUG. A partir de un valor inicial del RTG de aproximadamente 13 mmol/L, en los estudios en Fase 1, se observó supresión máxima del RTG promedio de 24 horas con la dosis de 300 mg diaria a aproximadamente 4 a 5 mmol/L en pacientes con diabetes tipo 2, lo que sugiere un riesgo bajo de hipoglucemia inducida por el tratamiento. Las reducciones en el RTG condujeron a un aumento de la EUG en sujetos con diabetes tipo 2 tratados con canagliflozina 100 mg o 300 mg que oscila desde 77 y 119 g/día en todos los estudios en fase 1; la EUG observada se traduce a una pérdida de 308 a 476 kcal/día. Las reducciones en el RTG y los aumentos en la EUG se mantuvieron durante un período de dosificación de 26 semanas en pacientes con diabetes tipo 2. Se observaron aumentos moderados (generalmente < 400-500 mL) en el volumen diario de orina que se atenuaron a lo largo de varios días de la dosificación. La excreción del ácido úrico en la orina aumentó de manera transitoria por la canagliflozina (se incrementó en un 19% en comparación con el valor basal en el día 1 y luego atenuándose hasta 6% en el día 2 y 1% en el día 13). Esto fue acompañado por una reducción sostenida de la concentración sérica de ácido úrico de aproximadamente el 20%.
Electrofisiología cardíaca: En un estudio aleatorizado, doble ciego, controlado con placebo, con comparador activo, cruzado de 4 vías, se administró a 60 sujetos sanos una dosis oral única de canagliflozina 300 mg, canagliflozina 1200 mg (4 veces la dosis recomendada máxima), moxifloxacino y placebo. No se observaron cambios significativos en el intervalo QTc con la dosis recomendada de 300 mg o con la dosis de 1200 mg. Con la dosis de 1200 mg, las concentraciones plasmáticas máximas de canagliflozina fueron aproximadamente 1.4 veces las concentraciones máximas en el estado estacionario después de una dosis de 300 mg una vez al día.
Eficacia Clínica: La co-administración de canagliflozina y metformina se ha estudiado en pacientes con diabetes tipo 2 inadecuadamente controlada con la dieta y el ejercicio y como complemento a la terapia con otros agentes antihiperglucémicos.
No se han realizado estudios de eficacia clínica con VOKANAMET™; sin embargo, la bioequivalencia de VOKANAMET™ con canagliflozina y metformina co-administradas como comprimidos individuales se demostró en sujetos sanos.
Canagliflozina: Canagliflozina se ha estudiado como un complemento a la terapia con metformina, sulfonilurea, metformina y sulfonilurea, metformina y una tiazolidinediona (pioglitazona), como un complemento a la terapia con insulina (con o sin otros agentes antihiperglucémicos, incluyendo metformina) y como monoterapia. La eficacia de canagliflozina fue comparada a un inhibidor DPP-4 (sitagliptina) y a una sulfonilurea (glimepirida). Canagliflozina fue también evaluada en pacientes mayores y pacientes con enfermedad cardiovascular o con alto riesgo de enfermedad cardiovascular.
Un total de 10285 pacientes con diabetes tipo 2 participaron en nueve estudios de eficacia y seguridad clínica, doble ciego, controlados realizados para evaluar los efectos de la canagliflozina sobre el control glucémico, incluyendo 5151 pacientes tratados con canagliflozina en combinación con metformina. La distribución racial de los pacientes que recibieron canagliflozina fue 72% Blancos, 16% asiáticos, 4% negros y 8% otros grupos. Aproximadamente el 16% de los pacientes fueron Hispanos. Aproximadamente el 58% de los pacientes fueron varones. Los pacientes tenían una edad promedio general de 59.6 años (rango de 21 a 96 años), con 3082 pacientes de 65 años de edad y mayores y 510 pacientes ≥ 75 años de edad. Además, un estudio doble ciego, controlado con placebo, de Fase 2 de 18 semanas con dosis de dos veces al día (canagliflozina 50 mg o 150 mg en combinación con metformina 500 mg) se realizó en 279 pacientes de los cuales 186 pacientes fueron tratados con canagliflozina en combinación con metformina.
En pacientes con diabetes tipo 2, el tratamiento con canagliflozina produjo mejorías clínica y estadísticamente significativas en la A1C, glucosa plasmática en ayunas (GPA) y glucosa postprandial (GPP) en dos horas, en comparación con el placebo. La canagliflozina fue efectiva en reducir la A1C en un amplio rango de pacientes independientemente de la duración de la enfermedad y el uso concomitante de agentes antihiperglucémicos para tratar la diabetes tipo 2. Se observaron mejorías estadísticamente significativas en el control glicémico con canagliflozina respecto al placebo cuando fue administrada como complemento a la terapia inicial con metformina, complemento a la terapia con metformina y sulfonilurea, metformina y pioglitazona o como complemento a la terapia con insulina (con o sin otros agentes antihiperglucémicos, incluyendo metformina). Además, se observaron mejorías significativas en la A1C con canagliflozina en los pacientes mayores. Las reducciones en la A1C fueron observadas en los subgrupos incluyendo la edad, género, raza, índice de masa corporal basal (BMI) y función basal de las células beta. Se observaron mayores reducciones en la A1C respecto al placebo en pacientes con A1C o valores de la TFGe basales más altos (ver sección Propiedades Farmacocinéticas).
Complemento a la terapia con metformina: Un total de 1284 pacientes con control glucémico inadecuado (A1C de ≥7% a ≤10.5%) con monoterapia con metformina (2000 mg/día o por lo menos 1500 mg/día si la dosis más elevada no es tolerada) participaron en un estudio clínico, multicéntrico, aleatorizado, doble ciego, controlado con placebo y activo, de 4 grupos, de grupo paralelo para evaluar la eficacia de canagliflozina como un complemento a la terapia con metformina durante 26 semanas. La edad promedio fue 55 años, el 47% de los pacientes fueron varones y la TFGe basal promedio fue 89 mL/min/1.73 m2. Los pacientes ya con metformina (N=1009) en el análisis con control glucémico inadecuado completaron un período de experimentación con placebo simple ciego de 2 semanas. Otros pacientes con metformina y otro agente oral o uno menor a la dosis requerida de metformina (N=275) fueron cambiados a un régimen de monoterapia con metformina. Después de por lo menos 8 semanas con una dosis estable de monoterapia con metformina, los pacientes ingresaron a un período de experimentación con placebo simple ciego de 2 semanas. Los pacientes fueron aleatorizados para la adición de canagliflozina 100 mg, canagliflozina 300 mg, sitagliptina 100 mg o el placebo, administrados una vez al día.
Como se muestra en el Tabla 6, se observaron mejorías estadísticamente significativas (p<0.001) en la A1C, GPA, GPP, peso corporal y presión sanguínea sistólica respecto al placebo. Además, un mayor porcentaje de pacientes alcanzaron una A1C < 7.0% en comparación con el placebo. Un número menor de pacientes con canagliflozina en combinación con metformina requirieron terapia de rescate glucémico: 1.6% de los pacientes con canagliflozina 100 mg, 0.3% de los pacientes con canagliflozina 300 mg y 14.8% de los pacientes con el placebo.
|
Tabla 6. Resultados de un estudio clínico controlado con placebo de canagliflozina como complemento a la terapia con metformina1 |
|||
|
Parámetro de eficacia |
Canagliflozina + Metformina 26 semanas |
Placebo + Metformina (N=183) |
|
|
100 mg (N=368) |
300 mg (N=367) |
||
|
A1C (%) |
|||
|
Valor basal (promedio) |
7.94 |
7.95 |
7.96 |
|
Cambio desde el valor basal (promedio ajustado) |
-0.79 |
-0.94 |
-0.17 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-0.622 (-0.76; -0.48) |
-0.772 (-0.91; -0.64) |
N/A3 |
|
Porcentaje de pacientes que alcanzaron A1C < 7% |
45.52 |
57.82 |
29.8 |
|
Glucosa plasmática en ayunas (mmol/L) |
|||
|
Valor basal (promedio) |
9.36 |
9.59 |
9.12 |
|
Cambio desde el valor basal (promedio ajustado) |
-1.52 |
-2.10 |
0.14 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-1.652 (-1.99; -1.32) |
-2.232 (-2.57; -1.90) |
N/A3 |
|
Glucosa postprandial de 2 horas (mmol/L) |
|||
|
Valor basal (promedio) |
14.30 |
14.54 |
13.81 |
|
Cambio desde el valor basal (promedio ajustado) |
-2.66 |
-3.17 |
-0.55 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-2.122 (-2.73; -1.51) |
-2.622 (-3.24; -2.01) |
N/A3 |
|
Peso Corporal |
|||
|
Valor basal (promedio) en Kg |
88.7 |
85.4 |
86.7 |
|
% del cambio desde el valor basal (promedio ajustado) |
-3.7 |
-4.2 |
-1.2 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-2.52 (-3.1; -1.9) |
-2.92 (-3.5; -2.3) |
N/A3 |
|
Presión sanguínea sistólica (mmHg) |
|||
|
Valor basal (promedio) |
128.0 |
128.7 |
128.0 |
|
Cambio desde el valor basal (promedio ajustado) |
-3.8 |
-5.1 |
1.5 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-5.42 (-7.3; -3.4) |
-6.62 (-8.5; -4.6) |
N/A3 |
|
1 Población con intención de tratar utilizando la última observación en el estudio previo a la terapia de rescate glucémico. 2 p<0.001 en comparación con el placebo. 3 N/A = No aplica. |
|||
Estudio controlado con activo frente a glimepirida como complemento a la terapia con metformina: Un total de 1450 pacientes con control glucémico inadecuado (nivel A1C de ≥7% a ≤9.5%) con monoterapia con metformina (≥ 2000 mg/día o por lo menos 1500 mg/día si la dosis más alta no es tolerada) participaron en un estudio clínico multicéntrico, aleatorizado, doble ciego, de 3 grupos, de grupo paralelo, controlado con activo para evaluar la eficacia de canagliflozina como complemento a la terapia con metformina durante 52 semanas. La edad promedio fue 56 años, el 52% de los pacientes fueron varones y la TFGe basal promedio fue 90 mL/min/1.73 m2. Los pacientes con metformina (N=928) a una dosis estable especificada en el protocolo ingresaron a un período de experimentación con placebo simple ciego de 2 semanas. Otros pacientes (N=522) ingresaron a un periodo de valoración de dosis de metformina y estabilización de la dosis/eliminación del agente antihiperglucémico, inmediatamente seguido por del período de experimentación de 2 semanas. Después del periodo de experimentación, los pacientes con control glucémico inadecuado fueron aleatorizados para la adición de canagliflozina 100 mg, canagliflozina 300 mg o glimepirida (valoración permitida durante el estudio de 52 semanas a 6 a 8 mg), administrados una vez al día.
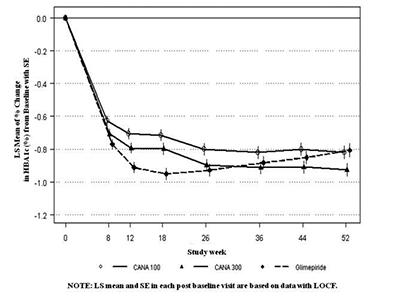
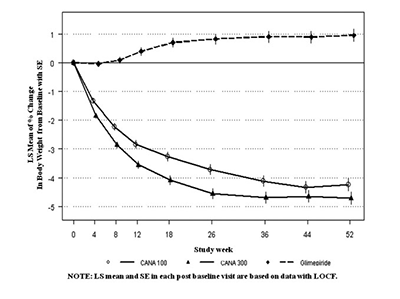
Como se muestra en el Tabla 7 y Figura 1, después de las 52 semanas, el tratamiento con canagliflozina 100 mg proporcionó reducciones similares en la A1C desde el valor basal en comparación con glimepirida (con el límite superior del intervalo de confianza al 95% alrededor de la diferencia entre grupos menos que el margen de no inferioridad pre-especificado de 0.3%); canagliflozina 300 mg proporcionó una reducción superior (p<0.05) desde el valor basal de la A1C en comparación con glimepirida (con el límite superior del intervalo de confianza al 95% debajo de 0). Se observaron mejorías estadísticamente significativas (p<0.001) en el peso corporal con canagliflozina en comparación con glimepirida. La incidencia de hipoglucemia con canagliflozina fue significativamente menor (p<0.001) en comparación con glimepirida. Un número menor de pacientes con canagliflozina requirieron terapia de rescate glucémico: 6.6% de los pacientes con canagliflozina 100 mg, 4.9% de los pacientes con canagliflozina 300 mg y 10.6% de los pacientes con glimepirida.
Un subconjunto de pacientes (N=208) quienes se sometieron a DXA y a exploraciones abdominales CT para la evaluación de la composición corporal demostraron que aproximadamente dos tercios del peso perdido con canagliflozina se debió a la pérdida de masa grasa con cantidades similares de grasa subcutánea visceral y abdominal perdida.
|
Tabla 7. Resultados de un estudio clínico de 52 semanas comparando canagliflozina con glimepirida como complemento a la terapia con metformina1 |
|||
|
Parámetro de Eficacia |
Canagliflozina + Metformina 52 semanas |
Glimepirida (valorada) + Metformina (N=482) |
|
|
100 mg (N=483) |
300 mg (N=485) |
||
|
A1C (%) |
|||
|
Valor basal (promedio) |
7.78 |
7.79 |
7.83 |
|
Cambio desde el valor basal (promedio ajustado) |
-0.82 |
-0.93 |
-0.81 |
|
Diferencia con glimepirida (promedio ajustado) (IC al 95%) |
-0.012 (-0.11; 0.09) |
-0.122 (-0.22; -0.02) |
N/A3 |
|
Porcentaje de pacientes que alcanzaron A1C < 7% |
53.6 |
60.1 |
55.8 |
|
Glucosa plasmática en ayunas (mmol/L) |
|||
|
Valor basal (promedio) |
9.18 |
9.09 |
9.20 |
|
Cambio desde el valor basal (promedio ajustado) |
-1.35 |
-1.52 |
-1.02 |
|
Diferencia con glimepirida (promedio ajustado) (IC al 95%) |
-0.33 (-0.56; -0.11) |
-0.51 (-0.73; -0.28) |
N/A3 |
|
Peso corporal |
|||
|
Valor basal (promedio) |
86.8 |
86.6 |
86.6 |
|
% del cambio desde el valor basal (promedio ajustado) |
-4.2 |
-4.7 |
1.0 |
|
Diferencia con glimepirida (promedio ajustado) (IC al 95%) |
-5.24 (-5.7; -4.7) |
-5.74 (-6.2; -5.1) |
N/A3 |
|
Presión sanguínea sistólica (mmHg)5 |
|||
|
Valor basal (promedio) |
130.0 |
130.0 |
129.5 |
|
Cambio desde el valor basal (promedio ajustado) |
-3.3 |
-4.6 |
0.2 |
|
Diferencia con glimepirida (promedio ajustado) (IC al 95%) |
-3.5 (-4.9; -2.1) |
-4.8 (-6.2; -3.4) |
N/A3 |
|
1 Población con intención de tratar utilizando la última observación en el estudio previo a la terapia de rescate glucémico. 2 Cumplió el criterio pre-especificado para no inferioridad a glimepirida (con límite superior del IC al 95% alrededor de la diferencia entre grupos menos que el margen de no inferioridad pre-establecido de < 0.3%). En una evaluación pre-especificada, el límite superior del IC al 95% para canagliflozina 300 mg, pero no para canagliflozina 100 mg fue < 0, indicando una reducción superior (p<0.05) en la A1C respecto a glimepirida con canagliflozina 300 mg. 3 N/A = No aplica 4 p<0.001 5 Incluye solo pacientes que tuvieron valores basales y post-basales. |
|||
Figura 1.
Cambios promedio desde el valor basal para A1C (%) y peso corporal durante 52 semanas en un estudio comparando canagliflozina con glimepirida como complemento a la terapia con metformina.
Complemento a la terapia con metformina y sulfonilurea: Un total de 469 pacientes con control glucémico inadecuado (nivel A1C de ≥7% a ≤10.5%) con combinación de metformina (2000 mg/día o por lo menos 1500 mg/día si la dosis más alta no es tolerada) y sulfonilurea (dosis máxima o casi máxima efectiva) participaron en un estudio clínico multicéntrico, aleatorizado, doble ciego, controlado con placebo, de 3 grupos, de grupo paralelo para evaluar la eficacia de canagliflozina como complemento a la terapia con metformina y sulfonilurea durante 26 semanas. La edad promedio fue 57 años, el 51% de los pacientes fueron varones y TFGe basal promedio fue 89 mL/min/1.73 m2. Los pacientes con dosis efectivas máximas o casi máximas de metformina y sulfonilurea (N=372) ingresaron a un período de experimentación con placebo simple ciego de 2 semanas. Otros pacientes (N=97) ingresaron a un periodo de valoración de dosis de metformina y sulfonilurea y estabilización de la dosis/eliminación del agente antihiperglucémico de hasta 12 semanas, seguido inmediatamente por el período de experimentación de 2 semanas. Después del periodo de experimentación, los pacientes con control glucémico inadecuado fueron aleatorizados para la adición de canagliflozina 100 mg, canagliflozina 300 mg o el placebo, administrados una vez al día. Como se muestra en la Tabla 8, se observaron mejorías estadísticamente significativas (p<0.001) en la A1C, GPA y peso corporal respecto al placebo. Además, un porcentaje mayor de pacientes alcanzaron una A1C < 7.0% en comparación con el placebo. Un número menor de pacientes con canagliflozina requirieron terapia de rescate glucémico: 1.3% de los pacientes con canagliflozina 100 mg, 1.9% de los pacientes con canagliflozina 300 mg y 12.8% de los pacientes con el placebo. Se observó una incidencia incrementada de hipoglucemia en este estudio, consistente con el incremento esperado de hipoglucemia cuando un agente no asociado con hipoglucemia es adicionado a sulfonilurea (ver sección Advertencias y Precauciones y Reacciones Adversas).
|
Tabla 8. Resultados de un estudio clínico controlado con placebo de 26 semanas de canagliflozina como complemento a la terapia con metformina y sulfonilurea1 |
|||
|
Parámetro de Eficacia |
Canagliflozina + Metformina y Sulfonilurea 26 semanas |
Placebo + Metformina y Sulfonilurea (N=156) |
|
|
100 mg (N=157) |
300 mg (N=156) |
||
|
A1C (%) |
|||
|
Valor basal (promedio) |
8.13 |
8.13 |
8.12 |
|
Cambio desde el valor basal (promedio ajustado) |
-0.85 |
-1.06 |
-0.13 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-0.712 (-0.90; -0.52) |
-0.922 (-1.11; -0.73) |
N/A3 |
|
Porcentaje de pacientes que alcanzaron A1C < 7% |
43.22 |
56.62 |
18.0 |
|
Glucosa plasmática en ayunas (mmol/L) |
|||
|
Valor basal (promedio) |
9.60 |
9.34 |
9.42 |
|
Cambio desde el valor basal (promedio ajustado) |
-1.01 |
-1.69 |
0.23 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-1.242 (-1.75; -0.73) |
-1.922 (-2.43; -1.41) |
N/A3 |
|
Peso Corporal |
|||
|
Valor basal (promedio) en Kg |
93.5 |
93.5 |
90.8 |
|
% de cambio desde el valor basal (promedio ajustado) |
-2.1 |
-2.6 |
-0.7 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-1.42 (-2.1; -0.7) |
-2.02 (-2.7; -1.3) |
N/A3 |
|
Presión Sanguínea Sistólica (mmHg) |
|||
|
Valor basal (promedio) |
130.4 |
130.8 |
130.1 |
|
Cambio desde el valor basal (promedio ajustado) |
-4.9 |
-4.3 |
-2.6 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-2.2 (-4.7; 0.2) |
-1.6 (-4.1; 0.9) |
N/A3 |
|
1 Población con intención de tratar utilizando la última observación en el estudio previo a la terapia de rescate glucémico. 2 p<0.001 en comparación con el placebo. 3 N/A = No aplica o no fue medido en este estudio. |
|||
Estudio controlado con activo frente a sitagliptina como complemento a la terapia con metformina y sulfonilurea: Un total de 755 pacientes con control glucémico inadecuado (nivel A1C de ≥7.0% a ≤10.5%) con la combinación de metformina (2000 mg/día o por lo menos 1500 mg/día si la dosis más alta no es tolerada) y sulfonilurea (dosis máxima o casi máxima efectiva) participaron en un estudio clínico multicéntrico, doble ciego, controlado con activo, de 2 grupos, de grupo paralelo para evaluar la eficacia de canagliflozina 300 mg como complemento a la terapia con metformina y sulfonilurea frente a sitagliptina 100 mg como complemento a la terapia con metformina y sulfonilurea durante 52 semanas. La edad promedio fue 57 años, el 56% de los pacientes fueron varones y la TFGe basal promedio fue 88 mL/min/1.73 m2. Los pacientes con dosis máximas o casi máximas efectivas de metformina y sulfonilurea (N=716) ingresaron a un período de experimentación con placebo simple ciego de 2 semanas. Otros pacientes (N=39) ingresaron a un periodo de valoración de dosis de metformina y sulfonilurea y dosis de estabilización de hasta 12 semanas, inmediatamente seguido por un período de experimentación de 2 semanas. Luego del periodo de experimentación, los pacientes con control glucémico inadecuado fueron aleatorizados para la adición de canagliflozina 300 mg o sitagliptina 100 mg.
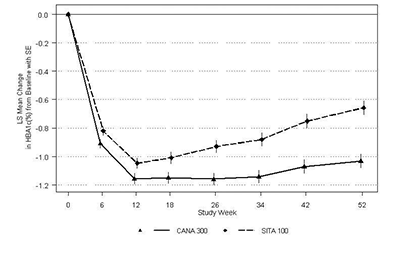
Como se muestra en la Tabla 9 y Figura 2, después de 52 semanas, canagliflozina 300 mg proporcionó una reducción superior (p<0.05) en la A1C en comparación con sitagliptina 100 mg (con el límite superior del intervalo de confianza al 95% alrededor de la diferencia entre grupos debajo de 0). Además, un porcentaje mayor de pacientes alcanzaron una A1C de < 7.0% con canagliflozina 300 mg respecto a sitagliptina: 47.6% de pacientes con canagliflozina 300 mg y 35.3% de los pacientes con sitagliptina. Los pacientes tratados con canagliflozina 300 mg mostraron una disminución significativa promedio en el cambio porcentual desde el valor basal del peso corporal en comparación con los pacientes que recibieron sitagliptina 100 mg. Se observó una incidencia incrementada similar de hipoglucemia con canagliflozina 300 mg y sitagliptina en este estudio, consistente con el incremento esperado de hipoglucemia cuando agentes no asociados con hipoglucemia son adicionados a sulfonilurea (ver sección Advertencias y Precauciones y Reacciones Adversas). La proporción de pacientes que cumplieron con el criterio de retiro glucémico (en base a GPA hasta la semana 26 y A1C posteriormente) fue menor con canagliflozina 300 mg (10.6%) en comparación con sitagliptina 100 mg (22.5%).
|
Tabla 9. Resultados del estudio clínico de 52 semanas comparando canagliflozina con sitagliptina como complemento a la terapia con metformina y sulfonilurea1 |
||
|
Parámetro de Eficacia |
Canagliflozina 300 mg + Metformina y Sulfonilurea (N=377) |
Sitagliptina 100 mg + Metformina y Sulfonilurea (N=378) |
|
A1C (%) |
||
|
Valor basal (promedio) |
8.12 |
8.13 |
|
Cambio desde el valor basal (promedio ajustado) |
-1.03 |
-0.66 |
|
Diferencia con sitagliptina (promedio ajustado) (IC al 95%) |
-0.372 (-0.50; -0.25) |
N/A4 |
|
Porcentaje de pacientes que alcanzaron A1C < 7% |
47.6 |
35.3 |
|
Glucosa plasmática en ayunas (mmol/L) |
||
|
Valor basal (promedio) |
9.42 |
9.09 |
|
Cambio desde el valor basal (promedio ajustado) |
-1.66 |
-0.32 |
|
Diferencia con sitagliptina (promedio ajustado) (IC al 95%) |
-1.34 (-1.66; -1.01) |
N/A4 |
|
Peso Corporal |
||
|
Valor basal (promedio) en Kg |
87.6 |
89.6 |
|
% de cambio desde el valor basal (promedio ajustado) |
-2.5 |
0.3 |
|
Diferencia con sitagliptina (promedio ajustado) (IC al 95%) |
-2.83 (-3.3; -2.2) |
N/A4 |
|
Presión sanguínea sistólica (mmHg) |
||
|
Valor basal (promedio) |
131.2 |
130.1 |
|
Cambio desde el valor basal (promedio ajustado) |
-5.1 |
0.9 |
|
Diferencia con sitagliptina (promedio ajustado) (IC al 95%) |
-5.93 (-7.6; -4.2) |
N/A4 |
|
1 Población con intención de tratar utilizando última observación en el estudio previo a la terapia de rescate glucémico. 2 Cumplió el criterio pre-especificado para no inferioridad a la sitagliptina (con límite superior del IC al 95% alrededor de la diferencia entre grupos menos que el margen de no inferioridad pre- especificado de < 0.3%); en una evaluación pre-especificada, el límite superior del IC al 95% para canagliflozina 300 mg fue < 0, indicando una reducción superior (p<0.05) en la A1C respecto a sitagliptina con canagliflozina 300 mg. 3 p<0.001. 4 N/A = No aplica. |
||
Figura 2:
Cambio promedio desde el valor basal para la A1C (%) durante 52 semanas en un estudio comparando canagliflozina con sitagliptina como complemento a la terapia con metformina y sulfonilurea.
Complemento a la terapia con metformina y pioglitazona: Un total de 342 pacientes con control glucémico inadecuado (nivel A1C de ≥7.0% a ≤10.5%) con combinación de metformina (2000 mg/día o por lo menos 1500 mg/día si la dosis más alta no es tolerada) y pioglitazona (30 o 45 mg/día) participaron en un estudio clínico multicéntrico, aleatorizado, doble ciego, de 3 grupos, de grupo paralelo, controlado con placebo para evaluar la eficacia de canagliflozina como complemento a la terapia con metformina y pioglitazona durante 26 semanas. La edad promedio fue 57 años, el 63% de los pacientes fueron varones y la TFGe basal promedio fue 86 mL/min/1.73 m2. Los pacientes ya con las dosis de metformina y pioglitazona especificadas en el protocolo (N=163) ingresaron a un período de experimentación con placebo simple ciego de 2 semanas. Otros pacientes (N=181) ingresaron a un periodo de valoración de dosis de metformina y pioglitazona y estabilización de dosis hasta de 12 semanas con dosis estables de metformina y pioglitazona por lo menos por 8 semanas, seguidas inmediatamente por un período de experimentación de 2 semanas. Después del período de experimentación, los pacientes con control glucémico inadecuado fueron aleatorizados (N=344) para la adición de canagliflozina 100 mg, canagliflozina 300 mg o el placebo, administrados una vez al día. Como se muestra en la Tabla 10, se observaron mejorías estadísticamente significativas (p<0.001) en la A1C, GPA y el peso corporal respecto al placebo para canagliflozina en la semana 26. Además, un porcentaje mayor de pacientes alcanzaron una A1C de < 7.0% en comparación con el placebo. Un número menor de pacientes con canagliflozina requirieron terapia de rescate glucémico: 0.9% de los pacientes con canagliflozina 100 mg, 0.0% de los pacientes con canagliflozina 300 mg y 12.2% de los pacientes con el placebo.
|
Tabla 10. Resultados de un estudio clínico controlado con placebo de 26 semanas de canagliflozina como complemento a la terapia con metformina y pioglitazona1 |
|||
|
Parámetro de Eficacia |
Canagliflozina + Metformina y Pioglitazona 26 semanas |
Placebo + Metformina y Pioglitazona (N=115) |
|
|
100 mg (N=113) |
300 mg (N=114) |
||
|
A1C (%) |
|||
|
Valor basal (promedio) |
7.99 |
7.84 |
8.00 |
|
Cambio desde el valor basal (promedio ajustado) |
-0.89 |
-1.03 |
-0.26 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-0.622 (-0.81; -0.44) |
-0.762 (-0.95; -0.58) |
N/A3 |
|
Porcentaje de pacientes que alcanzaron A1C < 7% |
46.92 |
64.32 |
32.5 |
|
Glucosa plasmática en ayunas (mmol/L) |
|||
|
Valor basal (promedio) |
9.38 |
9.11 |
9.13 |
|
Cambio desde el valor basal (promedio ajustado) |
-1.49 |
-1.84 |
0.14 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-1.632 (-2.05; -1.21) |
-1.982 (-2.41; -1.56) |
N/A3 |
|
Peso Corporal |
|||
|
Valor basal (promedio) en Kg |
94.2 |
94.4 |
94 |
|
% de cambio desde el valor basal (promedio ajustado) |
-2.8 |
-3.8 |
-0.1 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-2.72 (-3.6; -1.8) |
-3.72 (-4.6; -2.8) |
N/A3 |
|
Presión sanguínea sistólica (mmHg) |
|||
|
Valor basal (promedio) |
126.4 |
126.7 |
128.2 |
|
Cambio desde el valor basal (promedio ajustado) |
-5.3 |
-4.7 |
-1.2 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-4.1 (-6.9; -1.3) |
-3.5 (-6.3; -0.6) |
N/A3 |
|
1 Población con intención de tratar utilizando la última observación en el estudio previo a la terapia de rescate glucémico. 2 p<0.001 en comparación con el placebo 3 N/A = No aplica o no fue medido en este estudio. |
|||
Terapia combinada complementaria con metformina e inhibidores de la dipeptidil-peptidasa-4: Un total de 213 pacientes con diabetes tipo 2 no controlados adecuadamente con la combinación de metformina (mayor o igual a 1500 mg/día) y sitagliptina 100 mg/día (o una combinación de dosis fija equivalente) participaron en un estudio doble ciego, controlado con placebo, de 26 semanas, para evaluar la eficacia y la seguridad de canagliflozina en combinación con metformina y sitagliptina. La edad promedio fue 57 años, el 57% de los pacientes fueron varones y la TFGe basal promedio fue 90.5 mL/min/1.73 m2. Los pacientes que ya estaban en las dosis de metformina y sitagliptina especificadas en el protocolo (N=213) ingresaron en un período de prueba de placebo 2 semanas, simple ciego. Después del periodo de adaptación, los pacientes fueron aleatorizados a canagliflozina 100 mg o placebo, administrados una vez al día como complemento a metformina y la sitagliptina. Se realizó un ajuste de dosis a 300 mg de canagliflozina ya en la semana 6 en pacientes que requerían control glucémico adicional que presentaban TFGe apropiada y que toleraron 100 mg de canagliflozina.
Al final del tratamiento, canagliflozina una vez al día produjo una mejoría estadísticamente significativa en la HbA1C (p<0.001) en comparación con el placebo cuando se adicionó a metformina y la sitagliptina. Canagliflozina una vez al día también produjo una mejoría estadísticamente significativa en la proporción de pacientes que alcanzaron una HbA1C menor al 7%, una reducción estadísticamente significativa de la glucosa plasmática en ayunas (GPA) y en el porcentaje de reducción del peso corporal en comparación con el placebo cuando se adicionó a metformina y sitagliptina (ver Tabla 11). Se observó un cambio promedio estadísticamente significativo (p<0.001) del valor basal de la presión arterial sistólica con respecto al placebo de -5.85 mmHg con canagliflozina una vez al día. Para los pacientes que tomaron canagliflozina, el porcentaje que experimentó un evento adverso o discontinuación debido a un evento adverso ocurrió en el 39.8% y 0.9%, respectivamente, en comparación con el placebo que ocurrió en el 44.4% y 2.8%, respectivamente.
|
Tabla 11. Resultados del estudio clínico, controlado con placebo, de 26 semanas de canagliflozina en combinación con metformina y sitagliptina* |
||
|
Parámetro de Eficacia |
Placebo + Metformina y Sitagliptina (N=106) |
Canagliflozina + Metformina y Sitagliptina (N=107) |
|
HbA1C (%) |
||
|
Valor basal (promedio) |
8.38 |
8.53 |
|
Cambio desde el valor basal (promedio ajustado) |
-0.01 |
-0.91 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%)† |
-0.89‡ (-1.19; -0.59) |
|
|
Porcentaje de pacientes que alcanzaron HbA1C < 7% |
12 |
32 |
|
Glucosa plasmática en ayunas (mg/dL) |
||
|
Valor basal (promedio) |
180 |
186 |
|
Cambio desde el valor basal (promedio ajustado) |
-3 |
-30 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%)† |
-27‡ |
|
|
Peso Corporal |
||
|
Valor basal (promedio) en Kg |
89.9 |
93.8 |
|
% de cambio desde el valor basal (promedio ajustado) |
-1.6 |
-3.4 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%)† |
-1.8‡ (-2.7; -0.9) |
|
|
* Población con intención de tratar † Promedio ajustado e IC son derivados de un modelo mixto para medidas repetidas ‡ p<0.001. |
||
Complemento a la terapia con insulina (con o sin otros agentes antihiperglucémicos): Un total de 1718 pacientes con control glucémico inadecuado (nivel A1C de ≥7.0 a ≤ 10.5%) con insulina ≥ 30 unidades/día o insulina complementaria a la terapia con otros agentes antihiperglucémicos participaron en un sub estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo, de 3 grupos, de grupo paralelo de un estudio cardiovascular; este sub estudio evaluó la eficacia de canagliflozina en combinación con insulina (con o sin otros agentes antihiperglucémicos) durante 18 semanas. La edad promedio fue 63 años, el 66% de los pacientes fueron varones y la TFGe basal promedio fue 75 mL/min/1.73 m2. Los pacientes en insulina basal, en bolo o basal/en bolo por lo menos durante 10 semanas ingresaron a un período de 2 semanas de experimentación con placebo simple ciego. Aproximadamente el 70% de los pacientes estuvieron con un régimen previo de insulina basal/en bolo. Después del periodo de experimentación, los pacientes fueron aleatorizados a canagliflozina 100 mg, canagliflozina 300 mg o el placebo, administrados una vez al día como complemento a la insulina. La dosis de insulina diaria promedio al inicio del estudio fue 83 unidades, el cual fue similar en los grupos de tratamiento.
Un subgrupo de 432 pacientes con control glucémico inadecuado recibió canagliflozina o el placebo en combinación con metformina y ≥ 30 unidades/día de insulina durante 18 semanas. Como se muestra en la Tabla 12, se observaron mejorías estadísticamente significativas (p≤0.001) en la A1C, GPA, peso corporal y presión sanguínea sistólica (solamente 300 mg) respecto al placebo. Además, un porcentaje mayor de pacientes alcanzaron una A1C < 7.0% en comparación con el placebo. La dosis de insulina diaria promedio al inicio del estudio fue 93 unidades, el cual fue similar en los grupos de tratamiento. Un número menor de pacientes con canagliflozina en combinación con metformina e insulina requirieron terapia de rescate glucémico: 3.6% de los pacientes con canagliflozina 100 mg, 2.7% de los pacientes con canagliflozina 300 mg y 6.2% de los pacientes con el placebo. Se observó una incidencia incrementada de hipoglucemia en este estudio, el cual es consistente con el incremento esperado de hipoglucemia cuando un agente no asociado con hipoglucemia es adicionado a la insulina (ver sección Advertencias y Precauciones y Reacciones Adversas).
|
Tabla 12. Resultados de un estudio clínico controlado con placebo de 18 Semanas de canagliflozina como complemento a la terapia con insulina ≥ 30 Unidades/Día1 |
|||
|
Parámetro de Eficacia |
Canagliflozina 100 mg + Metformina + Insulina (N=139) |
Canagliflozina 300 mg + Metformina + Insulina (N=148) |
Placebo + Insulina (N=145) |
|
A1C (%) |
|||
|
Valor basal (media) |
8.20 |
8.22 |
8.15 |
|
Cambio desde el valor basal (promedio ajustado) |
-0.64 |
-0.79 |
0.03 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-0.662 (-0.81; -0.51) |
-0.822 (-0.96; -0.67) |
N/A4 |
|
Porcentaje de pacientes que alcanzaron A1C < 7% |
193 |
292 |
9 |
|
Glucosa plasmática en ayunas (mmol/L) |
|||
|
Valor basal |
9.34 |
9.30 |
9.05 |
|
Cambio desde el valor basal (promedio ajustado) |
-0.87 |
-1.34 |
0.03 |
|
Diferencia con el placebo (promedio ajustado) (IC al 97.5%) |
-0.912 (-1.54; -0.28) |
-1.382 (-2.00;-0.76) |
N/A4 |
|
Peso Corporal |
|||
|
Valor basal (media) en Kg |
99.7 |
101.1 |
102.3 |
|
% de cambio desde el valor basal (promedio ajustado) |
-1.7 |
-2.7 |
0.0 |
|
Diferencia con el placebo (promedio ajustado) (IC al 97.5%) |
-1.72 (-2.4; -1.0) |
-2.72 (-3.4; -2.0) |
N/A4 |
|
Presión sanguínea sistólica (mmHg) |
|||
|
Valor basal (media) |
136.2 |
141.7 |
138.3 |
|
Cambio desde el valor basal (promedio ajustado) |
-5.2 |
-7.7 |
-1.7 |
|
Diferencia con el placebo (promedio ajustado) (IC al 97.5%) |
-3.5 (-6.9; -0.1) |
-6.02 (-9.4; -2.7) |
N/A4 |
|
1 Población con intención de tratar a utilizando la última observación en el estudio previo a la terapia de rescate glucémico. 2 p≤0.001 en comparación con el placebo. 3 p≤0.01 en comparación con el placebo. 4 N/A = No aplica. |
|||
Estudio en pacientes mayores: Un total de 714 pacientes mayores (≥55 a ≤80 años de edad) con control glucémico inadecuado (nivel basal A1C de ≥7.0 a ≤10.0%) con terapia actual para la diabetes (solo dieta y ejercicios o en combinación con agentes orales o parenterales) participaron en un estudio aleatorizado, doble ciego, controlado con placebo para evaluar la eficacia de canagliflozina como complemento a la terapia con tratamiento actual para la diabetes durante 26 semanas. Un subgrupo de 609 pacientes con control glucémico inadecuado recibió canagliflozina o el placebo en combinación con metformina. La edad promedio fue 64 años, el 55% de los pacientes fueron varones y la TFGe basal promedio fue 77 mL/min/1.73 m2. Los pacientes con control glucémico inadecuado con sus terapias actuales para la diabetes fueron aleatorizados para la adición de canagliflozina 100 mg, canagliflozina 300 mg o el placebo, administrados una vez al día. Como se muestra en el Tabla 13, se observaron cambios estadísticamente significativos (p<0.001) desde el valor basal de la A1C, GPA y el peso corporal para canagliflozina en la semana 26. Además, un porcentaje mayor de pacientes alcanzaron una A1C de < 7.0% en comparación con el placebo. Un número menor de pacientes con canagliflozina requirió terapia de rescate glucémico: 2.1% de los pacientes con canagliflozina 100 mg, 0.4% de los pacientes concanagliflozina 300 mg y 11.0% de los pacientes con placebo (ver sección Propiedades Farmacocinéticas – Poblaciones especiales).
Un subgrupo de pacientes (N=211) participó en el sub estudio de composición corporal utilizando el análisis de composición corporal DXA. Esto demostró que aproximadamente dos tercios del peso perdido con canagliflozina se debió a la pérdida de la masa grasa respecto al placebo. No hubo cambios significativos en la densidad ósea en las regiones trabeculares y corticales.
|
Tabla 13. Resultados de un estudio clínico controlado con placebo de 26 Semanas de canagliflozina como complemento a la terapia con agentes antihiperglucémicos en pacientes mayores inadecuadamente controlados con agentes antihiperglucémicos (AHAs)1 |
|||
|
Parámetro de Eficacia |
Canagliflozina + AHA Actual 26 Semanas |
Placebo + AHA Actual N=237 |
|
|
100 mg N=241 |
300 mg N=236 |
||
|
A1C (%) |
|||
|
Valor basal (promedio) |
7.77 |
7.69 |
7.76 |
|
Cambio desde el valor basal (promedio ajustado) |
-0.60 |
-0.73 |
-0.03 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-0.572 (-0.71; -0.44) |
-0.702 (-0.84; -0.57) |
N/A3 |
|
Porcentaje de pacientes que logran A1C < 7% |
47.72 |
58.52 |
28.0 |
|
Glucosa plasmática en ayunas (mmol/L) |
|||
|
Valor basal (promedio) |
8.93 |
8.49 |
8.68 |
|
Cambio desde el valor basal (promedio ajustado) |
-1.00 |
-1.13 |
0.41 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-1.412 (-1.76; -1.07) |
-1.542 (-1.88; -1.19) |
N/A3 |
|
Peso Corporal |
|||
|
Valor basal (promedio) en Kg |
88.4 |
88.8 |
91.3 |
|
% de cambio desde el valor basal (promedio ajustado) |
-2.4 |
-3.1 |
-0.1 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%) |
-2.32 (-2.8; -1.7) |
-3.02 (-3.5; -2.4) |
N/A3 |
|
Presión sanguínea sistólica (mmHg) |
|||
|
Valor basal (promedio) |
130.6 |
131.1 |
131.4 |
|
Cambio desde el valor basal (promedio ajustado)2 |
-3.5 |
-6.8 |
1.1 |
|
Diferencia con el placebo (promedio ajustado) (IC al 95%)2 |
-4.62 (-6.9; -2.4) |
-7.92 (-10.1; -5.6) |
N/A3 |
|
1 Población con intención de tratar utilizando la última observación en el estudio previo a la terapia de rescate glucémico. 2 p<0.001 en comparación con el placebo. 3 N/A = No aplica. |
|||
Comorbilidades Diabéticas:
Canagliflozina:
Presión sanguínea: En un análisis de 4 estudios controlados con placebo de 26 semanas (N=2313), se observaron reducciones promedio de la presión sanguínea sistólica respecto al placebo con canagliflozina 100 mg (-3.9 mmHg), canagliflozina 300 mg (-5.3 mmHg) y el placebo (-0.1 mmHg) independientemente del uso de medicamentos antihipertensivos en la basal. En esta misma población, hubo un pequeño efecto sobre la presión sanguínea diastólica con cambios promedio de -2.1 mmHg con canagliflozina 100 mg, -2.5 mmHg con canagliflozina 300 mg y -0.3 mmHg con el placebo, independientemente del uso de medicamentos antihipertensivos al en la basal. No hubo cambio discernible en la frecuencia cardíaca.
Efectos en los lípidos: En un análisis integrado de cuatro estudios controlados con placebo de 26 semanas, los pacientes con diabetes tipo 2 tratados con ambas dosis de canagliflozina tuvieron niveles séricos incrementados de colesterol total, LDL-C, y HDL-C (colesterol asociado a lipoproteínas de alta densidad) en comparación con cambios pequeños en el placebo, mientras que los niveles séricos de los triglicéridos disminuyeron en comparación con el placebo (ver Tabla 14). En la semana 26, la proporción LDL-C/HDL-C cambió mínimamente en comparación al valor basal en todos los tres grupos de tratamiento. Similar a los cambios de no-HDL-C, apolipoproteína B y el número de partículas de LDL-C (medidos en dos estudios) se incrementaron en menor medida en la monoterapia y en la terapia de combinación de metformina de 26 semanas en comparación a los cambios de LDL-C (ver sección Reacciones Adversas).
|
Tabla 14. Efecto de canagliflozina en mediciones de los lípidos en cuatro estudios controlados con placebo de 26 semanas1 |
|||
|
Canagliflozina 100 mg (N=833) |
Canagliflozina 300 mg (N=834) |
Placebo (N=646) |
|
|
Colesterol Total |
|||
|
Promedio basal (mediana) en mmol/L |
4.89 (4.81) |
4.81 (4.73) |
4.96 (4.87) |
|
Cambio promedio de mínimos cuadrados (mediana) en mmol/L |
0.10 (0.10) |
0.18 (0.21) |
-0.02 (-0.04) |
|
% de cambio promedio de mínimos cuadrados (mediana) de colesterol total |
3.4 (2.0) |
5.2 (4.7) |
0.9 (-0.8) |
|
LDL-C |
|||
|
Promedio basal (mediana) en mmol/L |
2.76 (2.74) |
2.70 (2.64) |
2.83 (2.74) |
|
Cambio promedio de mínimos cuadrados (mediana) en mmol/L |
0.06 (0.05) |
0.15 (0.15) |
-0.06 (-0.05) |
|
% de cambio promedio de mínimos cuadrados (mediana) de LDL-C |
5.7 (2.0) |
9.3 (6.0) |
1.3 (-2.3) |
|
HDL-C |
|||
|
Promedio basal (mediana) en mmol/L |
1.19 (1.14) |
1.20 (1.16) |
1.17 (1.14) |
|
Cambio promedio de mínimos cuadrados (mediana) en mmol/L |
0.09 (0.08) |
0.11 (0.11) |
0.03 (0.05) |
|
% de cambio promedio de mínimos cuadrados (mediana) de HDL-C |
9.4 (7.8) |
10.3 (9.6) |
4.0 (3.5) |
|
No-HDL-C |
|||
|
Promedio basal (mediana) en mmol/L |
3.70 (3.60) |
3.61 (3.52) |
3.79 (3.70) |
|
Cambio promedio de mínimos cuadrados (mediana) en mmol/L |
-0.00 (-0.01) |
0.07 (0.08) |
-0.06 (-0.08) |
|
% de cambio promedio de mínimos cuadrados (mediana) de no-HDL-C |
2.2 (-0.3) |
4.3 (2.0) |
0.7 (-2.4) |
|
Proporción LDL-C/HDL-C |
|||
|
Promedio basal (mediana) |
2.5 (2.4) |
2.4 (2.3) |
2.5 (2.4) |
|
Cambio promedio de mínimos cuadrados (mediana) |
-0.1 (-0.1) |
-0.1 (-0.1) |
-0.2 (-0.1) |
|
% de cambio promedio de mínimos cuadrados (mediana) % de cambio de la proporción |
-1.4 (-5.2) |
0.8 (-2.1) |
-0.8 (-6.5) |
|
Triglicéridos |
|||
|
Promedio basal (mediana) en mmol/L |
2.06 (1.73) |
2.04 (1.70) |
2.10 (1.85) |
|
Cambio promedio de mínimos cuadrados (mediana) en mmol/L |
-0.11 (-0.10) |
-0.22 (-0.13) |
-0.00 (-0.03) |
|
% de cambio promedio de mínimos cuadrados (mediana) de triglicéridos |
2.4 (-6.0) |
0.0 (-9.2) |
7.6 (-2.2) |
|
1 Como monoterapia o complemento a la terapia con metformina, metformina y sulfonilurea y metformina y pioglitazona. |
|||
Glucosa plasmática en ayunas: En cuatro estudios controlados con placebo, el tratamiento con canagliflozina como monoterapia o complemento a la terapia con uno o dos fármacos antihiperglucémicos orales resultó en cambios promedio desde el valor basal respecto al placebo en GPA de -1.2 mmol/L a -1.9 mmol/L para canagliflozina 100 mg y -1.9 mmol/L a -2.4 mmol/L para canagliflozina 300 mg, respectivamente. Estas reducciones fueron sostenidas durante el período de tratamiento y cerca del máximo después del primer día de tratamiento.
Glucosa post-prandial: Utilizando una prueba estandarizada de tolerancia a la comida mixta, se midió la glucosa postprandial en tres estudios clínicos controlados con placebo como monoterapia o complemento a la terapia con uno o dos fármacos antihiperglucémicos orales. La canagliflozina resultó en reducciones promedio de los cambios desde el valor basal respecto al placebo de glucosa postprandial de -1.5 mmol/L a -2.7 mmol/L para canagliflozina 100 mg y -2.1 mmol /L a -3.5 mmol/L para canagliflozina 300 mg, respectivamente, debido a las reducciones en la concentración de glucosa previa a la comida y excursiones reducidas de la glucosa post-prandial.
Función de las Células Beta: Los estudios clínicos en un subconjunto de pacientes con diabetes tipo 2 (N=297) con canagliflozina durante 26 semanas indican mejoría de la función de las células beta basada en mediciones tales como evaluación del modelo de homeostasis para la función de las células beta (HOMA2-%B) y mejoría de la tasa de secreción de insulina con pruebas de tolerancia a la comida mixta.
Metformina: El estudio prospectivo aleatorizado (UKPDS) ha establecido el beneficio a largo plazo del control intensivo de la glucosa en sangre en la diabetes tipo 2. El análisis de los resultados para pacientes con sobrepeso tratados con metformina después del fracaso de la dieta sola mostró:
• Una reducción significativa del riesgo absoluto de cualquier complicación relacionada con la diabetes en el grupo de metformina (29.8 eventos/1000 paciente–años) frente a la dieta sola (43.3 eventos/1000 paciente–años), p=0.0023, y frente a sulfonilurea combinada y los grupos de monoterapia de insulina (40.1 eventos/1000 paciente –años), p=0.0034.
• Una reducción significativa del riesgo absoluto de cualquier mortalidad relacionada con diabetes: Metformina 7.5 eventos/1000 paciente–años, dieta sola 12.7 eventos/1000 paciente-años, p=0.017.
• Una reducción significativa del riesgo absoluto de la mortalidad general: Metformina 13.5 eventos/1000 paciente –años frente a la dieta sola 20.6 eventos/1000 paciente –años, (p=0.011), y frente a sulfonilurea combinada y los grupos de monoterapia de insulina 18.9 eventos/1000 paciente –años (p=0.021).
• Una reducción significativa en el riesgo absoluto de infarto de miocardio: Metformina 11 eventos/1000 paciente–años, dieta sola 18 eventos/1000 paciente–años, (p=0.01).]
REACCIONES ADVERSAS: En esta sección, se presentan las reacciones adversas. Las reacciones adversas son eventos adversos que fueron considerados estar razonablemente asociados con el uso de canagliflozina en base a la evaluación exhaustiva de la información disponible del evento adverso. No se puede establecer con fiabilidad una relación causal con canagliflozina y metformina en casos individuales. Además, debido a que los ensayos clínicos se llevan a cabo bajo condiciones muy variables, las tasas de reacción adversa observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas de los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica clínica.
Canagliflozina: La seguridad de la canagliflozina se evaluó en 10285 pacientes con diabetes tipo 2, incluyendo 5151 pacientes tratados con canagliflozina en combinación con metformina. Además, se realizó un estudio, doble ciego, controlado con placebo de Fase 2 de 18 semanas con dosis dos veces al día (canagliflozina 50 mg o 150 mg como complemento a la terapia con 500 mg de metformina) en 279 pacientes en el cual 186 pacientes fueron tratados con canagliflozina como complemento a la terapia con metformina.
Se realizaron análisis de seguridad en pacientes que recibieron canagliflozina como complemento a la terapia con metformina (con o sin otros agentes antihiperglucémicos), como complemento a la terapia con insulina (con o sin otros agentes antihiperglucémicos, incluyendo metformina), como complemento a la terapia con sulfonilurea y canagliflozina como monoterapia. Cinco estudios controlados con placebo y activo investigaron canagliflozina como complemento a la terapia con otros agentes antihiperglucémicos: dos con metformina (26 y 52 semanas); dos con metformina y sulfonilurea (26 y 52 semanas) y uno con metformina y pioglitazona (26 semanas). Un estudio controlado con placebo investigó el uso de canagliflozina como complemento a la terapia con el régimen del tratamiento actual para la diabetes (incluyendo la metformina) en pacientes mayores. Estudios cardiovasculares se completaron en pacientes con diabetes tipo 2; se realizaron análisis de seguridad en dos subestudios controlados con placebo (18 semanas) a partir de este estudio cardiovascular que investigó canagliflozina en combinación con sulfonilurea y con insulina. Se estudió canagliflozina como monoterapia en un estudio controlado con placebo de 26 semanas de duración.
Se realizó la evaluación principal de la seguridad y la tolerabilidad en un análisis conjunto (N = 2313) de cuatro estudios clínicos controlados con placebo de 26 semanas (monoterapia y complemento a la terapia con metformina, metformina y sulfonilurea, y metformina y pioglitazona). Las reacciones adversas comunicadas con más frecuencia durante el tratamiento (≥ 5%) fueron candidiasis vulvovaginal, infecciones del tracto urinario y poliuria o polaquiuria. Las reacciones adversas que llevaron a la discontinuación del ≥ 0.5% de todos los pacientes tratados con canagliflozina en estos estudios fueron candidiasis vulvovaginal (0.7% de las mujeres) y balanitis o balanopostitis (0.5% de los varones). Se realizaron análisis adicionales de seguridad (incluyendo los datos a largo plazo) a través de todo el programa de canagliflozina (estudios controlados con placebo y activo) para evaluar los eventos adversos reportados con la finalidad de identificar las reacciones adversas.
La Tabla 3 incluye las reacciones adversas reportadas en el ≥ 2% de los pacientes tratados con canagliflozina en los cuatro estudios clínicos en conjunto, controlados con placebo, de 26 semanas (N = 2313), incluyendo 1275 pacientes con la combinación de canagliflozina y metformina.
|
Tabla 3. Reacciones adversas de cuatro estudios en conjunto, controlados con placebo de 26 semanas1 reportadas en ≥ 2% de los pacientes tratados con canagliflozina |
|||
|
Clasificación por órganos y sistemas Reacción adversa |
Canagliflozina 100 mg N = 833 % |
Canagliflozina 300 mg N = 834 % |
Placebo N = 646 % |
|
Trastornos gastrointestinales |
|||
|
Estreñimiento |
15 (1.8) |
19 (2.3) |
6 (0.9) |
|
Náuseas |
18 (2.2) |
19 (2.3) |
10 (1.5) |
|
Sed2 |
23 (2.8) |
19 (2.3) |
1 (0.2) |
|
Trastornos renales y urinarios |
|||
|
Poliuria o polaquiuria3 |
44 (5.3) |
38 (4.6) |
5 (0.8) |
|
Infección del tracto urinario4 |
49 (5.9) |
36 (4.3) |
26 (4.0) |
|
Trastornos del sistema reproductor y de la mama |
|||
|
Balanitis o balanopostitis5 |
17 (4.2) |
15 (3.7) |
2 (0.6) |
|
Candidiasis vulvovaginal6 |
44 (10.4) |
49 (11.4) |
10 (3.2) |
|
1 Incluye monoterapia y complemento a la terapía con metformina, metformina y sulfonilurea, y metformina y pioglitazona. 2 Sed incluye los términos sed (1.3%, 1.9%, 0.2%) con incidencias para canagliflozina 100 mg, canagliflozina 300 mg y el placebo, respectivamente, y también incluye los términos boca seca y polidipsia con incidencias < 1% en cualquiera de los grupos de tratamiento. 3 Poliuria o polaquiuria incluye los términos poliuria (0.7%, 1.4%, 0.0%) y polaquiuria (4.2%, 3.1%, 0.6%) con incidencias para canagliflozina 100 mg, canagliflozina 300 mg y el placebo, respectivamente, y también incluye los términos aumento de diuresis, urgencia miccional y Nocturia con incidencias < 1% en cualquiera de los grupos de tratamiento. 4 Infección del tracto urinario incluye el término Infección del tracto urinario (5.5%, 4.1%, 4.0%) con incidencias para canagliflozina 100 mg, canagliflozina 300 mg y el placebo, respectivamente, y también incluye los términos cistitis, infección del riñón y sepsis urológica con incidencias < 1% en cualquiera de los grupos de tratamiento. No hubo desequilibrios entre canagliflozina 100 mg, canagliflozina 300 mg y el placebo en infección del riñón o sepsis urológica. 5 Balanitis o balanopostitis incluye los términos balanitis (2.2%, 1.7%, 0.0%) y balanopostitis (1.0%, 0.7%, 0.3%), con incidencias para canagliflozina 100 mg, canagliflozina 300 mg y el placebo, respectivamente, también incluye los términos balanitis candidiásica e Infección genital micótica con incidencias < 1% en cualquiera de los grupos de tratamiento. 6 Candidiasis vulvovaginal incluye los términos candidiasis vulvovaginal (1.6%, 2.8%, 1.0%) e Infección vulvovaginal micótica (5.9%, 5.3%, 1.3%), vulvovaginitis (1.9%, 1.6%, 0.0%) e infección vaginal (1.2%, 1.6%, 0.6%) con incidencias para canagliflozina 100 mg, canagliflozina 300 mg y placebo, respectivamente, y también incluye los términos vulvitis e infección genital micótica con incidencias < 1% en cualquiera de los grupos de tratamiento. |
|||
Otras reacciones adversas en estudios clínicos de canagliflozina que ocurrieron con una tasa < 2% en estudios controlados con placebo fueron reacciones adversas relacionadas al volumen intravascular reducido (mareo postural, hipotensión ortostática, hipotensión, deshidratación y síncope) (ver abajo), erupciones cutáneas y urticaria.
Descripción de reacciones adversas seleccionadas:
Cetoacidosis diabética: Se identificó CAD como una reacción adversa durante la vigilancia posterior a la comercialización. En una revisión de los datos del programa de desarrollo clínico para diabetes mellitus tipo 2, las tasas de incidencia de eventos adversos graves de CAD, cetoacidosis, acidosis metabólica y acidosis fueron de 0.0522 (0.07%, 4/5337), 0.0763 (0.11%, 6/5350) y 0.0238 (0.03%, 2/6909) por 100 pacientes-años con 100 mg de canagliflozina, 300 mg de canagliflozina y el comparador, respectivamente. De los 10 pacientes con canagliflozina, 6 (3 con 100 mg de canagliflozina, 3 con 300 mg de canagliflozina) fueron reportados por tener diabetes autoinmune (diabetes autoinmune latente del adulto [LADA, por sus siglas en inglés] o diabetes tipo 1) o dieron positivo para anticuerpos GAD65 mientras que ningún paciente con el comparador fue diagnosticado con diabetes autoinmune y 8 de los 10 pacientes estaban recibiendo terapia con insulina. Los valores de glucosa en sangre en 9 pacientes con canagliflozina alrededor de la hora de admisión oscilaron desde 19.3 mmol/L [347 mg/dL] hasta 31.7 mmol/L [571 mg/dL]. Un paciente tuvo valores de glucosa en sangre que oscilaron desde 8.2 mmol/L [148 mg/dL] hasta 17.8 mmol/L [320 mg/dL] (ver sección Advertencias y Precauciones).
Amputación del miembro inferior: En pacientes con diabetes tipo 2 que tenían enfermedad cardiovascular establecida o al menos dos factores de riesgo para enfermedad cardiovascular, canagliflozina se asoció con un incremento de aproximadamente 2 veces el riesgo de amputación del miembro inferior según lo observado en el Programa Integrado CANVAS compuesto por CANVAS y CANVAS-R, dos estudios clínicos grandes, a largo plazo, aleatorizados, controlados con placebo en los que se evaluó a 10142 pacientes. El desequilibrio ocurrió tempranamente a las primeras 26 semanas de la terapia. Se hizo seguimiento a los pacientes en CANVAS y CANVAS-R durante un promedio de 5.7 y 2.1 años, respectivamente. Independientemente del tratamiento con canagliflozina o el placebo, el riesgo de amputación fue mayor en pacientes con antecedente de amputación previa en el estado basal, enfermedad vascular periférica y neuropatía. El riesgo de amputación del miembro inferior no fue dependiente de la dosis. Los resultados de amputación para el Programa Integrado CANVAS se muestran en la Tabla 4. En los otros estudios de diabetes tipo 2 con canagliflozina, en los cuales se enroló a una población de 8114 pacientes diabéticos, no se observó diferencia en el riesgo de amputación del miembro inferior en comparación con el grupo control.
|
Tabla 4. Análisis integrado de las amputaciones en CANVAS y CANVAS-R |
||
|
Placebo N=4344 |
Canagliflozina N=5790 |
|
|
Número total de sujetos con eventos, n (%) |
47 (1.1) |
140 (2.4) |
|
Tasa de incidencia (por 100 sujetos-años) |
0.34 |
0.63 |
|
HR (IC del 95%) frente al placebo |
1.97 (1.41, 2.75) |
|
|
Amputación menor, n (%)* |
34/47 (72.3) |
99/140 (70.7) |
|
Amputación mayor, n (%)† |
13/47 (27.7) |
41/140 (29.3) |
|
Nota: La incidencia se basa en el número de pacientes con al menos una amputación, y no en el número total de eventos de amputación. El seguimiento del paciente se calcula a partir del día 1 hasta la fecha del primer evento de amputación. Algunos pacientes presentaron más de una amputación. * Dedo del pie y parte media del pie. † Tobillo, por debajo de la rodilla y encima de la rodilla. |
||
De los sujetos que presentaron un evento de amputación, el dedo del pie y la porción media del pie fueron los sitios más frecuentes (71%) en ambos grupos de tratamiento (ver Tabla 4). Las amputaciones múltiples (algunos involucraron ambos miembros inferiores) se observaron con poca frecuencia y en proporciones similares en ambos grupos de tratamiento.
Las infecciones del miembro inferior, úlceras del pie diabético, enfermedad arterial periférica y gangrena fueron los eventos médicos más comunes asociados con la necesidad de una amputación en ambos grupos de tratamiento (ver sección Advertencias y precauciones).
Volumen intravascular reducido: En el análisis agrupado de los cuatro estudios controlados con placebo de 26 semanas, la incidencia de todas las reacciones adversas relacionadas con el volumen intravascular reducido (por- ejemplo, mareo postural, hipotensión ortostática, hipotensión, deshidratación y síncope) fue del 1.2% para canagliflozina 100 mg, 1.3% para canagliflozina 300 mg y 1.1% para el el placebo. La incidencia con el tratamiento con canagliflozina en los dos estudios controlados con activo fue similar a los comparadores.
En el estudio cardiovascular específico, donde los pacientes eran generalmente mayores con una prevalencia más elevada de comorbilidades, las incidencias de reacciones adversas relacionadas con el volumen intravascular reducido fueron del 2.8% con canagliflozina 100 mg, 4.6% con canagliflozina 300 mg y 1.9% con el placebo.
Para evaluar los factores de riesgo de estas reacciones adversas, se realizó un análisis conjunto más amplio (N = 9439) de pacientes de ocho estudios controlados de Fase 3 incluyendo dosis de canagliflozina de 100 mg y 300 mg. En este análisis conjunto, los pacientes con diuréticos del asa, los pacientes con insuficiencia renal moderada (TFGe 30 a < 60 mL/min/1,73 m2) (CrCl 30 a < 60 mL/min) y los pacientes ≥ 75 años de edad tuvieron mayores incidencias de estas reacciones adversas. En los pacientes con diuréticos del asa, las incidencias fueron 3.2% con canagliflozina 100 mg y 8.8% con canagliflozina 300 mg en comparación con el 4.7% en el grupo control. En los pacientes con una TFGe basal < 60 mL/min/1.73 m2 (CrCl 30 a < 60 mL/min), las incidencias fueron 4.8% con canagliflozina 100 mg y 8.1% con canagliflozina 300 mg en comparación con 2.6% en el grupo de control. En los pacientes ≥ 75 años de edad, las incidencias fueron 4.9% con canagliflozina 100 mg y 8.7% con canagliflozina 300 mg en comparación con 2.6% en el grupo control (ver sección Posología y Administración, Advertencias y precauciones y Propiedades Farmacocinéticas - Poblaciones Especiales).
En el estudio cardiovascular específico y en el análisis conjunto más amplio, las discontinuaciones debido a las reacciones adversas relacionadas con el volumen intravascular reducido y las reacciones adversas graves relacionadas con el volumen intravascular reducido no aumentaron con la canagliflozina.
Hipoglucemia en el tratamiento complementario con insulina o secretagogo de insulina: La frecuencia de hipoglucemia fue baja (< 6%) entre los grupos de tratamiento cuando se utilizó como monoterapia o como un complemento de los agentes antihiperglucémicos no asociados con la hipoglucemia. La hipoglucemia se reportó con más frecuencia cuando canagliflozina se usó en combinación con insulina o sulfonilurea, lo cual es consistente con el aumento esperado de hipoglucemia cuando un agente no asociado con hipoglucemia se adiciona a la insulina o a un secretagogo de insulina (por ejemplo, sulfonilurea). En un subestudio de 18 semanas, cuando se adicionó canagliflozina a la terapia con insulina, se observó hipoglucemia en 49.3%, 48.2% y 36.8% de los pacientes tratados con canagliflozina 100 mg, canagliflozina 300 mg y el placebo, respectivamente, y ocurrió hipoglucemia severa en 1.8%, 2.7% y 2.5% de los pacientes tratados con canagliflozina 100 mg, canagliflozina 300 mg y placebo, respectivamente. Cuando se adicionó canagliflozina a la terapia con sulfonilurea, se observó hipoglucemia en el 4.1%, 12.5% y 5.8% de los pacientes tratados con canagliflozina 100 mg, canagliflozina 300 mg y el placebo, respectivamente (ver sección Posología y Administración y Advertencias y Precauciones).
Infecciones micóticas genitales: Se reportó candidiasis vulvovaginal (incluyendo vulvovagitinis e infección micótica vulvovaginal) en 10.4% y 11.4% de las pacientes de sexo femenino tratadas con canagliflozina 100 mg y canagliflozina 300 mg, respectivamente, en comparación al 3.2% de las pacientes de sexo femenino tratadas con el placebo. La mayoría de los reportes de candidiasis vulvovaginal ocurrieron durante los primeros cuatro meses del tratamiento con canagliflozina. Entre las pacientes de sexo femenino que estaban tomando canagliflozina, el 2.3% experimentaron más de una infección. En general, el 0.7% de todas las pacientes de sexo femenino discontinuaron canagliflozina debido a candidiasis vulvovaginal (ver sección Advertencias y Precauciones).
Se reportó balanitis o balanopostitis candidiásica en el 4.2% y 3.7% de los pacientes de sexo masculino tratados con canagliflozina 100 mg y canagliflozina 300 mg, respectivamente, en comparación con el 0.6% de los pacientes de sexo masculino tratados con el placebo. Entre los pacientes de sexo masculino que estaban tomando canagliflozina, el 0.9% tuvo más de una infección. En general, el 0.5% de los pacientes de sexo masculino discontinuaron canagliflozina debido a balanitis o balanopostitis candidiásica. Se reportó fimosis en 0.3% de los varones no circuncidados en un análisis combinado de 8 ensayos controlados. En este análisis conjunto, también se reportó que el 0.2% de los pacientes de sexo masculino tratados con canagliflozina estaban circuncidados (ver sección Advertencias y Precauciones).
Infecciones del tracto urinario: Las infecciones del tracto urinario fueron reportadas con más frecuencia con canagliflozina 100 mg y 300 mg (5.9% frente a 4.3%, respectivamente) en comparación con 4.0% con el placebo. La mayoría de las infecciones fueron leves a moderadas sin aumentos en la ocurrencia de eventos adversos graves. Los sujetos respondieron a los tratamientos estándar mientras continuaban con el tratamiento con canagliflozina. La incidencia de infecciones recurrentes no se incrementó con canagliflozina.
Fractura ósea: En un estudio cardiovascular de 4327 pacientes con enfermedad cardiovascular conocida o de alto riesgo, las tasas de incidencia de fractura ósea fueron 16.3, 16.4, y 10.8 por cada 1000 pacientes-año de exposición a canagliflozina 100 mg, canagliflozina 300 mg y el placebo, respectivamente, con el desequilibrio de fractura ocurriendo inicialmente dentro de las primeras 26 semanas de terapia. En otros estudios de diabetes tipo 2 con canagliflozina, los cuales enrolaron a una población general con diabetes de aproximadamente 5800 pacientes, no se observó diferencia en el riesgo de fractura respecto al control. Después de 104 semanas de tratamiento, la canagliflozina no afectó de manera adversa la densidad mineral ósea.
Pruebas de laboratorio: Los valores de laboratorio, descritos abajo, son derivados del análisis conjunto de los estudios clínicos controlados con placebo, de 26 semanas, a menos que se indique lo contrario.
Aumento del potasio sérico: Los cambios porcentuales promedio desde el valor basal del potasio en sangre fueron 0.5% y 1.0% para la canagliflozina 100 mg y 300 mg, respectivamente, en comparación con 0.6% para el placebo. Se observaron episodios de potasio sérico elevado (> 5.4 mEq/L y 15% por encima del valor basal) en el 4.4% de los pacientes tratados con canagliflozina 100 mg, 7.0% de los pacientes tratados con canagliflozina 300 mg y 4.8% de los pacientes tratados con el placebo. En general, las elevaciones fueron leves (< 6.5 mEq/L), transitorias y no necesitaron tratamiento específico.
Aumento de la creatinina sérica y nitrógeno ureico en sangre: Los cambios porcentuales promedio desde el valor basal de la creatinina, con disminuciones proporcionales en la TFGe fueron 2.8% y 4.0% para canagliflozina 100 mg y 300 mg, respectivamente, en comparación con 1.5% para el placebo. El promedio de los cambios porcentuales desde la basal del nitrógeno ureico en sangre fue 17.1% y 18.0% para canagliflozina 100 mg y 300 mg, respectivamente, en comparación con 2.7% para el placebo. Se observaron estos cambios generalmente dentro de las seis semanas del inicio del tratamiento. Posteriormente, las concentraciones de creatinina sérica tendieron gradualmente hacia el valor basal y los niveles de nitrógeno ureico en sangre permanecieron estables.
La proporción de pacientes con mayores disminuciones de la TFGe (> 30%) desde el valor basal, ocurriendo en cualquier momento durante el tratamiento, fue 2.0% con canagliflozina 100 mg y 4.1% con canagliflozina 300 mg respecto al 2.1% con el placebo. Estas disminuciones de la TFGe fueron a menudo transitorias con un número menor de pacientes teniendo este nivel de disminución al final del estudio, ocurriendo en el 0.7% de los pacientes con canagliflozina 100 mg, 1.4% de los pacientes con canagliflozina 300 mg y 0.5% de los pacientes tratados con el placebo (ver sección Advertencias y Precauciones).
Estos cambios en los valores de laboratorio mejoraron o retornaron a los valores basales después de la discontinuación de la terapia con canagliflozina.
Cambios en los lípidos: Los cambios porcentuales promedio desde el valor basal respecto al placebo para el colesterol asociado a lipoproteínas de baja densidad (LDL-C) fueron de 0.11 mmol/L (4.4 mg/dL) (4.5%) y 0.21 mmol/L (8.2 mg/dL) (8.0%) con canagliflozina 100 mg y canagliflozina 300 mg, respectivamente. Se observaron aumentos más pequeños en el colesterol total de 2.5% y 4.3% respecto al placebo para canagliflozina 100 mg y canagliflozina 300 mg, respectivamente. Los aumentos en el colesterol asociado a lipoproteínas de alta densidad (HDL-C) fueron 5.4% y 6.3% con respecto al placebo para canagliflozina 100 mg y canagliflozina 300 mg, respectivamente. Los aumentos en el no –HDL-C respecto al placebo fueron de 0.05 mmol/L (1.5%) y 0.13 mmol/L (3.6%) con canagliflozina 100 mg y 300 mg, respectivamente. Las proporciones de LDL-C/HDL-C no cambiaron con las dosis de canagliflozina en comparación con el placebo. Las concentraciones de ApoB y del número de partícula LDL-C (medidas en dos estudios) y no-HDL-C aumentaron en menor medida en comparación con los cambios en el LDL-C.
Aumento de la hemoglobina: Los cambios promedio (cambios porcentuales) desde el valor basal de las concentraciones de hemoglobina fueron de 4.7 g/L (3.5%) con canagliflozina 100 mg, 5.1 g/L (3.8%) con canagliflozina 300 mg y -1.8 g/L (-1.1%) con el placebo. Se observaron pequeños incrementos proporcionales en el cambio porcentual promedio desde el valor de basal de los hematocritos y eritrocitos en la sangre. Al final del tratamiento, el 4.0%, 2.7% y 0.8% de los pacientes tratados con canagliflozina 100 mg, canagliflozina 300 mg y el placebo, respectivamente, tuvieron niveles de hemoglobina por encima del límite superior normal.
Aumento de fosfato sérico: Se observaron aumentos relacionados con la dosis de los niveles de fosfato sérico con la canagliflozina. En el conjunto de cuatro ensayos controlados con placebo, los cambios porcentuales promedio de los niveles de fosfato sérico fueron 3.6% y 5.1% con canagliflozina 100 mg y canagliflozina 300 mg, respectivamente, en comparación con el 1.5% con el placebo. Se observaron episodios de fosfato sérico elevado (> 1.65 mmol/L y 25% por encima del valor basal) en el 0.6% y 1.6% de los pacientes tratados con canagliflozina 100 mg y 300 mg, respectivamente, en comparación con 1.3% de los pacientes tratados con el placebo.
Disminución de urato sérico: Se observaron disminuciones moderadas en el cambio porcentual promedio desde el valor basal del urato sérico en los grupos de canagliflozina 100 mg y 300 mg (-10.1% y -10.6%, respectivamente) en comparación con el placebo, donde un ligero incremento (1.9%) desde el valor basal fue observado. Las disminuciones del urato sérico en los grupos de canagliflozina fueron máximas o casi máximas en la Semana 6 y se mantuvieron con la dosificación. Se observó un aumento transitorio de la excreción urinaria de ácido úrico, el cual no fue persistente. En un análisis conjunto (N = 9439) de los pacientes de ocho estudios controlados de Fase 3 incluyendo ambas dosis de canagliflozina, no se incrementaron los eventos de nefrolitiasis.
Seguridad cardiovascular: Se realizó un meta análisis prospectivo, pre-especificado de eventos cardiovasculares adjudicados independientemente a partir de los estudios clínicos de Fase 2 y 3 en 9632 pacientes con diabetes tipo 2, incluyendo 4327 pacientes quienes están participando en un estudio cardiovascular en curso (pacientes con enfermedad cardiovascular o con alto riesgo de enfermedad cardiovascular). El cociente de riesgo para el criterio de valoración principal (tiempo hasta el evento en el compuesto de muerte cardiovascular, accidente cerebrovascular no fatal, infarto de miocardio no fatal y angina inestable que requiere hospitalización) para canagliflozina (ambas dosis en conjunto) frente a comparadores combinados de activo y placebo fue de 0.91 (IC al 95%: 0.68, 1.22). Los cocientes de riesgo para las dosis de 100 mg y 300 mg fueron similares. Por lo tanto, no hubo evidencia de un aumento en el riesgo cardiovascular con canagliflozina 100 mg o canagliflozina 300 mg respecto a los comparadores.
Reacciones adversas en poblaciones específicas:
Pacientes ancianos: El perfil de seguridad en pacientes ancianos fue generalmente consistente con los pacientes más jóvenes. Los pacientes ≥ 75 años de edad tuvieron una mayor incidencia de reacciones adversas relacionadas al volumen intravascular reducido (tales como mareo postural, hipotensión ortostática, hipotensión) con incidencias de 4.9%, 8.7% y 2.6% con canagliflozina 100 mg, canagliflozina 300 mg y en el grupo control, respectivamente. Se reportaron disminuciones en la TFGe (-3.6% y -5.2%) con canagliflozina 100 mg y 300 mg, respectivamente, en comparación con el grupo control (-3.0%) (ver sección Posología y Administración y Advertencias y Precauciones).
Pacientes con una TFGe 45 a < 60 mL/min/1.73 m2 [CrCl 45 a < 60 mL/min]: En un análisis de pacientes con una TFGe basal de 45 a < 60 mL/min/1.73 m2 [CrCl 45 a < 60 mL/min], las incidencias de reacciones adversas relacionadas al volumen intravascular reducido fue 4.6% con canagliflozina 100 mg y 7.1% con canagliflozina 300 mg respecto al 3.4% con el placebo (ver sección Posología y Administración y Advertencias y Precauciones). Los niveles de creatinina sérica se incrementaron en un 4.9% y 7.3% con canagliflozina 100 mg y 300 mg, respectivamente, respecto al 0.2% con el placebo. Los niveles de nitrógeno ureico en sangre se incrementaron en un 13.2% y 13.6% con canagliflozina 100 mg y 300 mg, respectivamente, respecto al 0.7% con el placebo. La proporción de pacientes con mayores disminuciones de la TFGe (>30%) en cualquier momento durante del tratamiento fue 6.1%, 10.4% y 4.3% con canagliflozina 100 mg, canagliflozina 300 mg y el placebo, respectivamente. Al final del estudio, el 2.3% de los pacientes tratados con canagliflozina 100 mg, 4.3% con canagliflozina 300 mg y 3.5% con el placebo tuvieron ese tipo de disminuciones (ver sección Advertencias y Precauciones).
Las incidencias de potasio sérico elevado (> 5.4 mEq/L y 15% por encima del valor basal) fueron 5.2% con canagliflozina 100 mg, 9.1% con canagliflozina 300 mg y 5.5% con el placebo. Se observaron elevaciones más severas en raras ocasiones en pacientes con insuficiencia renal moderada quienes tuvieron previamente concentraciones elevadas de potasio y/o quienes estaban con medicamentos múltiples que reduce la excreción de potasio, como los diuréticos ahorradores de potasio y los inhibidores de la enzima convertidora de angiotensina (ECA). En general, las elevaciones fueron transitorias y no necesitaron tratamiento específico.
Los niveles de fosfato sérico se incrementaron en 3.3% y 4.2% con canagliflozina 100 mg y 300 mg, respectivamente, en comparación con 1.1% con el placebo. Las incidencias de fosfato sérico elevado (>1.65 mmol/L y 25% por encima del valor basal) fueron 1.4% con canagliflozina 100 mg, 1.3% con canagliflozina 300 mg y 0.4% con el placebo. En general, las elevaciones fueron transitorias y no necesitaron tratamiento específico.
Metformina: Las reacciones adversas más comunes debido al inicio de metformina son nauseas, vómitos, diarrea, dolor abdominal y pérdida de apetito.
El tratamiento a largo plazo con metformina ha sido asociado con la disminución de la vitamina B12, el cual muy pocas veces resulta en deficiencia clínicamente significativa de vitamina B12 (por ejemplo, anemia megaloblástica) (ver sección Advertencias y Precauciones).
Datos posteriores a la comercialización: Además de las reacciones adversas identificadas a partir de los estudios clínicos, las siguientes reacciones adversas han sido identificadas durante la experiencia posterior a la comercialización con canagliflozina. Debido a que estas reacciones fueron reportadas voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco. En la tabla, se proporcionan las frecuencias de acuerdo a la siguiente convención:
— Muy común: ≥ 1/10 (≥ 10%)
— Común: ≥ 1/100 y < 1/10 (≥ 1% y < 10%)
— Poco común: ≥ 1/1000 y < 1/100 (≥ 0.1% y < 1%)
— Rara: ≥ 1/10000 y < 1/1000 (≥ 0.01% y < 0.1%)
— Muy rara: < 1/10000, incluyendo reportes aislados (< 0.01%)
— No conocidas: No pueden estimarse a partir de los datos disponibles.
Canagliflozina:
|
Tabla 5. Reacciones adversas identificadas durante la experiencia posterior a la comercialización con canagliflozina |
||
|
Clasificación por órganos y sistemas Reacción Adversa |
Categoría de frecuencia Estimada a partir de las tasas de reportes espontáneos* |
Categoría de Frecuencia Estimada a partir de estudios clínicos |
|
Trastornos del metabolismo y la nutrición |
||
|
Cetoacidosis diabética |
Muy rara |
Rara |
|
Trastornos del sistema immune |
||
|
Reacción anafiláctica |
Muy rara |
Rara |
|
Trastornos de la piel y tejido subcutáneo |
||
|
Angioedema |
Muy rara |
Rara |
|
Trastornos renales y urinarios |
||
|
Pielonefritis |
Muy rara |
Rara |
|
Insuficiencia renal (relacionada con reducción del volumen) |
Muy rara |
Poco común |
|
Urosepsis |
Muy rara |
Rara |
|
*Las tasas de los reportes espontáneos posteriores a la comercialización se basaron en una exposición estimada de persona-año de tratamiento. |
||
INTERACCIONES: No se han realizado estudios de interacción farmacocinética con VOKANAMET™; sin embargo, tales estudios se han realizado con las sustancias activas individuales (canagliflozina y metformina) de VOKANAMET™.
La co-administración de canagliflozina (300 mg una vez por día) y de metformina (2000 mg una vez al día) no tuvo efecto clínicamente relevante en la farmacocinética de la canagliflozina ni de la metformina.
Canagliflozina:
Evaluación in vitro de las interacciones: El metabolismo de la canagliflozina es principalmente a través de la conjugación de glucorónido mediada por la transferasa glucoronosil UDP 1A9 (UGT1A9) y 2B4 (UGT2B4).
En los estudios in vitro, la canagliflozina no inhibió el citocromo P450 CYP1A2, CYP2A6, CYP2C19, CYP2D6 ni CYP2E1, CYP2B6, CYP2C8, CYP2C9, ni indujo a CYP1A2, CYP2C19, CYP2B6, CYP3A4 a concentraciones más elevadas que las terapéuticas. La canagliflozina inhibió débilmente a CYP3A4 in vitro; sin embargo, basado en un estudio clínico, no se observó interacción clínicamente relevante. Por lo tanto, no se espera que la canagliflozina altere la depuración metabólica de los medicamentos co-administrados que son metabolizados por estas enzimas.
La canagliflozina es un sustrato de glucoproteína P (gp-P) e inhibe el transporte mediado por la glucoproteína P de la digoxina con baja potencia.
Evaluación in vivo de las interacciones: Se realizaron estudios específicos de interacción de fármacos para investigar los efectos de los inhibidores o inductores de las enzimas metabolizadoras de fármacos UGT1A9 y UGT2B4 y de los transportadores gp-P y MRP2 sobre la farmacocinética de la canagliflozina, y también para evaluar los efectos de la canagliflozina sobre la farmacocinética del sustrato de gp-P digoxina.
Efectos de otros fármacos sobre canagliflozina: En los estudios clínicos, se evaluaron los efectos de otros fármacos sobre la canagliflozina. La ciclosporina, la hidroclorotiazida, los anticonceptivos orales (etinilestradiol y levonorgestrel), la metformina y el probenecid no tuvieron un efecto clínicamente relevante sobre la farmacocinética de la canagliflozina.
Rifampicina: La co-administración con rifampicina, un inductor no selectivo de varias enzimas UDP-glucuroniltransferasa (UGT) y transportadores de fármacos, incluyendo UGT1A9, UGT2B4, gp-P y MRP2, disminuyeron la exposición a canagliflozina. Estas disminuciones en la exposición a canagliflozina pueden disminuir la eficacia. Si un inductor combinado de estas UGT y sistemas de transporte de fármacos (por ejemplo, rifampicina, fenitoína, barbitúricos, fenobarbital, ritonavir, carbamazepina, efavirenz, hierba de San Juan [Hypericum perforatum]) deben coadministrarse con VOKANAMET™, monitorear la HbA1c en los pacientes que están recibiendo VOKANAMET™ conteniendo 50 mg de canagliflozina dos veces al día con la consideración de aumentar la dosis a VOKANAMET™ conteniendo 150 mg de canagliflozina dos veces al día si se requiere un control glucémico adicional. Se deben considerar otras terapias antihiperglucémicas en pacientes con una TFGe < 60 mL/min/1.73 m2 [CrCl <60 mL/min] que estan tomando VOKANAMET™ conteniendo 50 mg canagliflozina dos veces al día quienes están recibiendo terapia concomitante con un inductor de la enzima UGT y quienes requieren control glicémico adicional.
|
Tabla 1. Efectos de los fármacos co-administrados sobre la exposición sistémica de canagliflozina |
||||
|
Fármaco co-administrado |
Dosis del fármaco co-administrado1 |
Dosis de Canagliflozina1 |
Proporción geométrica promedio (Proporción con/sin fármaco co-administrado) Ningún efecto = 1.0 |
|
|
AUC2 (IC al 90%) |
Cmáx (IC al 90%) |
|||
|
No se requiere ajuste de la dosis de VOKANAMET™ para: |
||||
|
Ciclosporina |
400 mg |
300 mg una vez al día por 8 días |
1.23 (1.19; 1.27) |
1.01 (0.91; 1.11) |
|
Etinilestradiol y levonorgestrel |
0.03 mg de etinilestradiol y 0.15 mg de levonorgestrel |
200 mg una vez al día por 6 días |
0.91 (0.88; 0.94) |
0.92 (0.84; 0.99) |
|
Hidroclorotiazida |
25 mg una vez al día por 35 días |
300 mg una vez al día por 7 días |
1.12 (1.08; 1.17) |
1.15 (1.06; 1.25) |
|
Metformina |
2000 mg |
300 mg una vez al día por 8 días |
1.10 (1.05; 1.15) |
1.05 (0.96; 1.16) |
|
Probenecid |
500 mg dos veces al día por 3 días |
300 mg una vez al día por 17 días |
1.21 (1.16; 1.25) |
1.13 (1.00; 1.28) |
|
Rifampicina |
600 mg una vez al día por 8 días |
300 mg |
0.49 (0.44; 0.54) |
0.72 (0.61; 0.84) |
|
1Dosis única a menos que se indique lo contrario. 2AUCinf para fármacos administrados como dosis única y AUC24h para fármacos administrados como dosis múltiple. |
||||
Efectos de canagliflozina sobre otros fármacos:
En los estudios de interacción realizados en sujetos sanos, la canagliflozina en estado estacionario no tuvo efecto clínicamente relevante sobre la farmacocinética de la metformina, los anticonceptivos orales (etinil estradiol y levonorgestrel), la gliburida, la simvastatina, paracetamol, la hidroclorotiazida ni la warfarina.
Digoxina: La combinación de canagliflozina 300 mg una vez al día durante 7 días con una única dosis de 0.5 mg de digoxina seguida de 0.25 mg diarios durante 6 días resultó en un aumento del 20% en el AUC y un aumento del 36% en la Cmáx de digoxina, posiblemente debido a una interacción en el nivel de la gp-P. Los pacientes que están tomando digoxina u otros glicósidos cardíacos (por ejemplo, digitoxina) deben monitorearse apropiadamente.
|
Tabla 2: Efecto de canagliflozina sobre la exposición sistémica de los fármacos co-administrados |
|||||
|
Fármaco co-administrado |
Dosis del fármaco co-administrado1 |
Dosis de canagliflozina1 |
Proporción geométrica promedio (Proporción con/sin fármaco co-administrado) Ningún efecto = 1.0 |
||
|
AUC2 (IC al 90%) |
Cmáx (IC al 90%) |
||||
|
No se requiere ajuste de la dosis de VOKANAMET™ para: |
|||||
|
Digoxina |
0.5 mg una vez al día el primer día seguido por 0.25 mg una vez al día por 6 días |
300 mg una vez al día por 7 días |
digoxina |
1.20 (1.12; 1.28) |
1.36 (1.21; 1.53) |
|
Etinilestradiol y levonorgestrel |
0.03 mg de etinilestradiol y 0.15 mg de levonorgestrel |
200 mg una vez al día por 6 días |
etinilestradiol |
1.07 (0.99; 1.15) |
1.22 (1.10; 1.35) |
|
levonorgestrel |
1.06 (1.00; 1.13) |
1.22 (1.11; 1.35) |
|||
|
Gliburida |
1.25 mg |
200 mg una vez al día por 6 días |
gliburida |
1.02 (0.98; 1.07) |
0.93 (0.85; 1.01) |
|
3-cis-hidroxi- gliburida |
1.01 (0.96; 1.07) |
0.99 (0.91; 1.08) |
|||
|
4-trans-hidroxi- gliburida |
1.03 (0.97; 1.09) |
0.96 (0.88; 1.04) |
|||
|
Hidroclorotiazida |
25 mg una vez al día por 35 días |
300 mg una vez al día por 7 días |
hidroclorotiazida |
0.99 (0.95; 1.04) |
0.94 (0.87; 1.01) |
|
Metformina |
2000 mg |
300 mg una vez al día por 8 días |
metformina |
1.20 (1.08; 1.34) |
1.06 (0.93; 1.20) |
|
Paracetamol |
1000 mg |
300 mg dos veces al día por 25 días |
Paracetamol |
1.063 (0.98; 1.14) |
1.00 (0.92; 1.09) |
|
Simvastatina |
40 mg |
300 mg una vez al día por 7 días |
simvastatina |
1.12 (0.94; 1.33) |
1.09 (0.91; 1.31) |
|
simvastatina ácida |
1.18 (1.03; 1.35) |
1.26 (1.10; 1.45) |
|||
|
Warfarina |
30 mg |
300 mg una vez al día por 12 días |
(R) – warfarina |
1.01 (0.96; 1.06) |
1.03 (0.94; 1.13) |
|
(S) – warfarina |
1.06 (1.00; 1.12) |
1.01 (0.90; 1.13) |
|||
|
INR |
1.00 (0.98; 1.03) |
1.05 (0.99; 1.12) |
|||
|
1Dosis única a menos que se indique lo contrario. 2AUCinf para fármacos administrados como dosis única y AUC24h para fármacos administrados como dosis múltiple. 3AUC0-12h |
|||||
Interferencia con prueba de laboratorio/fármaco:
Ensayo 1,5-AG: Los aumentos en la excreción de glucosa urinaria con canagliflozina pueden reducir falsamente los niveles de 1,5-anhidroglucitol (1,5 AG) y hacer que las mediciones de 1,5 AG no sean confiables en la evaluación del control glucémico. Por lo tanto, los ensayos de 1,5-AG no deben utilizarse para la evaluación del control glucémico en pacientes con VOKANAMET™. Para más detalles, puede ser aconsejable contactar al fabricante específico del ensayo 1,5-AG.
Prueba de glucosa en orina: Debido al mecanismo de acción de la canagliflozina, los pacientes que toman VOKANAMET™ tendrán un resultado positivo en la prueba de glucosa en orina.
Metformina: Existe un mayor riesgo de acidosis láctica en intoxicación aguda con alcohol (particularmente en el caso de ayuno, desnutrición o insuficiencia hepática) debido al principio activo metformina de VOKANAMET™ (ver sección Advertencias y Precauciones). Debe evitarse el consumo de alcohol y de medicamentos que contengan alcohol.
Los fármacos catiónicos (por- ejemplo, amilorida, digoxina, morfina, procainamida, quinidina, quinina, ranitidina, triamtereno, trimetoprima o vancomicina) que son eliminados por secreción tubular renal, teóricamente tienen el potencial de interacción con la metformina compitiendo por los sistemas de transporte tubular renal comunes. Aunque tales interacciones todavía son teóricas (excepto para cimetidina), se recomienda un monitoreo cuidadoso del paciente y un ajuste de la dosis de VOKANAMET™ y/o del fármaco que produce la interferencia en pacientes que están tomando medicamentos catiónicos que son excretados a través del sistema de secreción tubular proximal renal.
La administración intravascular de agentes de contraste yodados en los estudios radiológicos puede conducir a insuficiencia renal, resultando en acumulación de metformina y en un riesgo de acidosis láctica. Por lo tanto, VOKANAMET™ debe discontinuarse antes, o en el momento de la prueba y no reinstituirla hasta 48 horas después, y solamente que se haya re-evaluado la función renal y ésta haya sido encontrada normal (ver sección Advertencias y Precauciones).
Los glucocorticoides (administrados por vía local y sistémica), los simpatomiméticos y los diuréticos tienen actividad hiperglucémica intrínseca. Se debe informar al paciente y deben realizarse monitoreos más frecuentes de la glucosa en sangre, especialmente al inicio del tratamiento con tales medicamentos. Si es necesario, la dosis del medicamento anti-hiperglucémico debe ajustarse durante la terapia con el otro medicamento y al discontinuarlo.
INFORMACIÓN NO CLÍNICA:
Canagliflozina: Los datos no clínicos no revelan riesgos particulares para los humanos basados en los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad y toxicidad sobre la reproducción y el desarrollo. En un estudio con ratas jóvenes se administró canagliflozina desde el día 21 hasta el día 90 postnatal no se demostró un incremento de la sensibilidad en comparación con los efectos observados en ratas adultas.
Carcinogenecidad y mutagenicidad: Canagliflozina no aumentó la incidencia de tumores en ratones machos y hembras en un estudio de dos años con dosis de 10, 30 y 100 mg/kg. La dosis más alta de 100 mg/kg proporcionó hasta 14 veces la dosis clínica de 300 mg basada en la exposición del AUC. Canagliflozina aumentó la incidencia de tumores testiculares de las células de Leydig en ratas macho en todas las dosis evaluadas (10, 30 y 100 mg/kg); la dosis más baja de 10 mg/kg es aproximadamente 1.5 veces la dosis clínica de 300 mg basada en la exposición del AUC. Las dosis más altas de canagliflozina (100 mg/kg) en ratas machos y hembras aumentaron la incidencia de feocromocitomas y tumores tubulares renales; basados en la exposición del AUC, el nivel de efecto no observable (NOEL) de 30 mg/kg/día para feocromocitomas y tumores renales tubulares es aproximadamente 4.5 veces la exposición a la dosis clínica diaria de 300 mg. Basados en estudios mecánicos preclínicos y clínicos, los tumores de las células de Leydig, los tumores de túbulos renales y feocromocitomas se consideraron específicos de la rata. Los tumores de túbulos renales inducidos por canagliflozina y los feocromocitomas en ratas parecen ser causados por mala absorción de carbohidratos como consecuencia de la actividad inhibitoria intestinal de SGLT1 de canagliflozina en el intestino de ratas; los estudios clínicos mecanísticos no han demostrado mala absorción de carbohidratos en humanos con dosis de canagliflozina de hasta 2 veces la dosis clínica máxima recomendada. Los tumores de células de Leydig son asociados con un aumento de la hormona luteinizante (LH), el cual es un mecanismo conocido de la formación de tumores de células de Leydig en ratas. En un estudio clínico de 12 semanas, la LH no estimulada no aumentó en los machos tratados con canagliflozina.
Canagliflozina no fue mutagénica con o sin activación metabólica en el ensayo de Ames. La canagliflozina fue mutagénica en el ensayo de linfoma de ratón in vitro, pero no sin activación metabólica. La canagliflozina no fue mutagénica o clastogénica en un ensayo oral de micronúcleo in vivo en ratas y en un ensayo de Comet oral in vivo en ratas.
Toxicología reproductiva: En un estudio de desarrollo pre y postnatal, la canagliflozina administrada a ratas hembra desde el Día 6 de la gestación hasta el día 20 de la lactancia resultó en una disminución del peso corporal en crías machos y hembra a dosis maternas tóxicas > 30 mg/kg/día (exposiciones ≥ 5.9 veces la exposición humana a canagliflozina con la dosis humana máxima recomendada [MRHD]). La toxicidad materna se limitó a la disminución del de peso corporal ganado.
En los estudios con ratas, la canagliflozina no tuvo efectos adversos sobre el desarrollo embrionario temprano, el apareamiento y la fertilidad hasta la dosis más alta de 100 mg/kg (hasta 19 veces la dosis clínica de 300 mg basada en la exposición del AUC).
Metformina: Los datos preclínicos no revelan riesgos especiales para humanos basados en los estudios convencionales de farmacología de seguridad, toxicidad de dosis repetidas, genotoxicidad, potencial carcinogénico o toxicidad para la reproducción.
Carcinogenicidad y mutagenicidad: Se han realizado estudios de carcinogenicidad a largo plazo en ratas (duración de la dosis de 104 semanas) y ratones (duración de la dosis de 91 semanas) con dosis de hasta 900 mg/kg/día y 1500 mg/kg/día, respectivamente. Estas dosis son aproximadamente 4 veces la dosis diaria humana máxima recomendada de 2000 mg basada en comparaciones del área de superficie corporal. No se encontró evidencia de carcinogenicidad con metformina en ratones machos o hembras. Del mismo modo, no hubo potencial tumorigénico observado con metformina en ratas macho. Sin embargo, hubo un aumento de la incidencia de pólipos uterinos estromales benignos en ratas hembra tratadas con 900 mg/kg/día.
No hubo evidencia de un potencial mutagénico de metformina en las siguientes pruebas in vitro: Prueba de Ames (S. typhimurium), prueba de mutación genética (células de linfoma de ratón) o prueba de aberraciones cromosómicas (linfocitos humanos). Los resultados en la prueba de micronúcleo de ratón in vivo también fueron negativos.
Fertilidad: La fertilidad de ratas macho o hembra no fue afectada por la metformina cuando se administró a dosis tan altas como 600 mg/kg/día, el cual es aproximadamente 3 veces la dosis máxima recomendada diaria en humanos basándose en las comparaciones del área de la superficie corporal.
ADVERTENCIAS Y PRECAUCIONES:
Lesión renal aguda y deterioro de la función renal: Considerar discontinuar temporalmente en condiciones de ingesta oral reducida o pérdida de líquidos. Si ocurre lesión renal aguda, discontinuar y tratarla inmediatamente. Monitorear la función renal durante la terapia.
General: No se ha establecido la seguridad y eficacia de VOKANAMET™ en pacientes con diabetes tipo 1. Se debe evitar el uso de VOKANAMET™ en estos pacientes.
Incremento del riesgo de fracturas óseas en pacientes que usan canagliflozina como principio activo único o en combinación.
Disminución en la densidad mineral ósea de la columna lumbar y de cadera en pacientes que usan canagliflozina como principio activo único o en combinación.
Acidosis láctica: La acidosis láctica es rara, pero grave, puede ocurrir complicación metabólica (mortalidad alta en ausencia de tratamiento rápido) debido a la acumulación de metformina. Han ocurrido casos reportados de acidosis láctica en pacientes con metformina principalmente en pacientes diabéticos con insuficiencia renal significativa. La incidencia de acidosis láctica puede y debe reducirse también evaluando otros factores de riesgo asociados tales como diabetes mal controlada, cetosis, ayuno prolongado, consumo excesivo de alcohol, insuficiencia hepática y cualquier condición asociada con hipoxia.
Diagnóstico: Debe considerarse el riesgo de acidosis láctica en el caso de signos no específicos, tales como calambres musculares, con trastornos digestivos, como dolor abdominal y astenia severa.
La acidosis láctica se caracteriza por disnea acidótica, dolor abdominal, hipotermia y coma. Los hallazgos del laboratorio de diagnóstico son pH sanguíneo disminuido, niveles de lactato en plasma superiores a 5 mmol/L y una diferencia aniónica y proporción lactato/piruvato aumentado. Si se sospecha acidosis metabólica, se debe discontinuar el tratamiento con el medicamento y hospitalizar al paciente inmediatamente (ver sección Sobredosis).
Advertencias y precauciones relacionas con el riesgo de presentar cetoacidosis: El riesgo de cetoacidosis diabética en pacientes tratados con inhibidores de SGLT2 debe considerarse ante la presencia de sintomatología inespecífica, como náuseas vómitos, dolor abdominal, anorexia, sed excesiva, disnea, confusión, o cansancio o somnolencia inusual, incluso con niveles de glucemia menores de 250 mg/dl.
Los factores que deben tenerse en cuenta al inicio y durante el tratamiento con un inhibidor de SGLT2, comprenden situaciones que pueden predisponer a la presentación de cetoacidosis diabética, como deshidratación, restricción de ingesta calórica, reducción de peso, infecciones, cirugía, vómitos, reducción de la dosis de insulina, mal control de la diabetes, o ingesta de alcohol; por lo cual estas situaciones deben tenerse en cuenta al prescribir un tratamiento con un inhibidor de SGLT2.
Si se sospecha el diagnóstico de cetoacidosis se debe suspender el tratamiento y realizar la determinación de cuerpos cetónicos. Los pacientes que hayan tenido cetoacidosis durante el tratamiento con inhibidores de SGLT2 no se les debería reintroducir el tratamiento, a no ser que otros factores hayan sido claramente los precipitantes y éstos se hayan resuelto.
En caso de pacientes hospitalizados por cirugía mayor o enfermedad médica grave, el tratamiento con inhibidores de SGLT2 debe interrumpirse hasta que se resuelva la situación.
Estos medicamentos se encuentran exclusivamente indicados para el tratamiento de la diabetes mellitus tipo 2.
Amputación del miembro inferior: En estudios clínicos a largo plazo de canagliflozina en pacientes con diabetes tipo 2 con enfermedad cardiovascular (ECV) establecida o con al menos 2 factores de riesgo para ECV, se ha observado un incremento de aproximadamente 2 veces el riesgo de amputación del miembro inferior (principalmente del dedo del pie y de la parte media del pie) en pacientes tratados con canagliflozina (ver sección Reacciones adversas). Dado que no se ha establecido un mecanismo subyacente, se desconocen los factores de riesgo de la amputación diferentes de los factores de riesgo generales.
Antes de iniciar el tratamiento con VOKANAMET™, considerar los factores en el antecedente del paciente que pueden incrementar el riesgo de amputación. Como medidas de precaución, se debe considerar realizar monitorización cuidadosa de los pacientes con un mayor riesgo de eventos de amputación y aconsejar a los pacientes sobre la importancia de los cuidados preventivos de rutina del pie y de mantener hidratación adecuada. También debe considerarse suspender el tratamiento con VOKANAMET™ en pacientes que desarrollan eventos que pueden preceder a la amputación, tales como úlceras en la piel, infección, osteomielitis o gangrena en la extremidad inferior.
Volumen intravascular reducido: Debido a su mecanismo de acción, la canagliflozina aumenta la excreción urinaria de glucosa (EUG) e induce una diuresis osmótica, lo cual puede reducir el volumen intravascular. Los pacientes más susceptibles a las reacciones adversas relacionadas al volumen intravascular reducido (por ejemplo, mareo postural, hipotensión ortostática o hipotensión) incluyen a los pacientes con diuréticos del asa, pacientes con insuficiencia renal moderada y pacientes ≥ 75 años de edad (ver sección Posología y Administración y Reacciones adversas).
En estudios clínicos de canagliflozina controlados con placebo, se observaron incrementos en las reacciones adversas relacionadas al volumen intravascular reducido comúnmente con la dosis de 300 mg y ocurrieron con mayor frecuencia en los primeros tres meses (ver sección Reacciones adversas).
Debido al volumen intravascular reducido, generalmente se observaron aumentos promedio pequeños dependientes de la dosis en la creatinina sérica y disminuciones concomitantes de la TFGe dentro de las primeras seis semanas del inicio del tratamiento con canagliflozina. En pacientes susceptibles a reducciones mayores del volumen intravascular como las descritas anteriormente, algunas veces se observaron mayores disminuciones de la TFGe (> 30%) las cuales subsecuentemente mejoraron y, con poca frecuencia necesitaron la interrupción del tratamiento con canagliflozina (ver sección Reacciones adversas).
Se debe aconsejar a los pacientes reportar síntomas de volumen intravascular reducido. Estas reacciones adversas con poca frececuencia llevaron a la discontinuación de canagliflozina y a menudo fueron manejados mediante la modificación del régimen del fármaco que reduce la presión sanguínea (incluyendo los diuréticos) mientras se continuaba con la tereapia con canagliflozina. En pacientes con reducción del volumen, se recomienda corregir esta condición antes del iniciar con VOKANAMET™.
Debe evaluarse la función renal antes de iniciar con VOKANAMET™. Se recomienda monitorear la función renal con más frecuecia en pacientes con TFGe < 60 mL/min/1.73 m2 (CrCl < 60 mL/min]). No debe utilizarse VOKANAMET™ en pacientes con TFGe < 60 ml/min/1,73 m2 (CrCl < 60 ml/min).
Función renal: Debido a que la metformina es excretada por los riñones, debe evaluarse la función renal antes de iniciar la terapia con VOKANAMET™ y por lo menos anualmente en lo sucesivo (ver sección Contraindicaciones). En pacientes con riesgo de desarrollar insuficiencia renal (por ejemplo, ancianos) o en pacientes con una TFGe < 60 mL/min/1.73 m2, la función renal debe evaluarse con mayor frecuencia.
VOKANAMET™ debe discontinuarse si se presenta evidencia de insuficiencia renal (por ejemplo, niveles de creatinina ≥ 1.5 mg/dL para varones o ≥ 1.4 mg/dL para mujeres o depuración anormal de creatinina). VOKANAMET™ no debe iniciarse en pacientes con una TFGe < 60 mL/min/1.73 m2 (CrCl < 60 mL/min) y debe discontinuarse cuando la TFGe sea persistentemente < 60 mL/min/1.73 m2 (CrCl < 60 mL/min).
La disminución de la función renal ocurre comúnmente en pacientes ancianos y puede ser asintomática. Se debe tener cuidado especial en situaciones donde la función renal puede deteriorarse, por ejemplo, al iniciar terapia antihipertensiva o diurética o al iniciar tratamiento con medicamentos antiinflamatorios no esteroideos (AINEs).
El/Los medicamento(s) concomitantes que pueden afectar la función renal o resultar en un cambio hemodinámico significante o interferir con la disposición de la metformina debe(n) utilizarse con cuidado (ver sección Interacciones y Propiedades Farmacológicas).
La canagliflozina como entidad única puede utilizarse en pacientes con TFGe de > 45 mL/min/ 1.73 m2 [CrCl > 45 mL/min] si VOKANAMET™ es inapropiado debido a la insuficiencia renal.
Administración de agente de contraste yodado: La administración intravascular de los agentes de contraste yodados en los estudios radiológicos puede conducir a insuficiencia renal, la cual ha sido asociada con acidosis láctica en pacientes que reciben metformina. Por lo tanto, VOKANAMET™ debe discontinuarse antes, o en el momento de la prueba y no restablecerse hasta 48 horas después y solamente después de que se haya re-evaluado la función renal y ésta haya sido encontrada normal (ver sección Interacciones).
Cirugía: Como VOKANAMET™ contiene clorhidrato de metformina, el tratamiento debe discontinuarse 48 horas antes de la cirugía electiva con anestesia general, espinal o epidural. VOKANAMET™ generalmente no debe reanudarse antes de 48 horas transcurridas y solamente después que se haya re-evaluado la función renal y ésta haya sido encontrada normal.
Cambio en el estado clínico de los pacientes con diabetes tipo 2 previamente controlada: Un paciente con diabetes tipo 2 previamente bien controlada con VOKANAMET™ que desarrolla anormalidades de laboratorio o enfermedades clínicas (especialmente enfermedades imprecisas y poco definidas) debe ser evaluado rápidamente para evidenciar cetoacidosis o acidosis láctica. La evaluación debe incluir electrolitos séricos y cetonas, glucosa en sangre y, si está indicado, los niveles de pH de la sangre, lactato, piruvato y metformina. Si ocurre acidosis de cualquier tipo, VOKANAMET™ debe interrumpirse inmediatamente e iniciarse otras medidas correctivas adecuadas.
Deterioro de la función hepática: Como se ha asociado deterioro de la función hepática con algunos casos de acidosis láctica con metformina, VOKANAMET™ generalmente debe evitarse en pacientes con evidencia clínica o de laboratorio de enfermedad hepática.
Hipoglucemia en terapia complementaria con insulina o secretagogos de insulina: La canagliflozina demostró tener una baja incidencia de hipoglucemia cuando se utilizó sola o como terapia complementaria con agentes antihiperglucémicos no asociados con hipoglucemia. La insulina y los secretagogos de insulina (por ejemplo, sulfonilurea) son conocidos por causar hipoglucemia. Cuando se utilizó canagliflozina como complemento a la terapia con insulina o un secretagogo de insulina (por ejemplo, sulfonilurea), la incidencia de hipoglucemia se incrementó por encima que la del placebo.
Por lo tanto, para reducir el riesgo de hipoglucemia, se puede considerar una reducción de la dosis de insulina o un secretagogo de insulina (ver sección Posología y Administración y Reacciones adversas).
Infecciones micóticas genitales: Consistente con el mecanismo de inhibición SGLT2 con aumento de la EUG, se reportaron candidiasis vulvovaginal en mujeres y balanitis o balanopostitis en varones en ensayos clínicos (ver sección Reacciones adversas). Los pacientes de sexo masculino y femenino con antecedente de infecciones micóticas genitales fueron más propensos de desarrollar una infección. La balanitis o balanopostitis ocurrieron principalmente en pacientes de sexo masculino no circuncidados; también se reportaron eventos de fimosis. En un análisis conjunto de 8 ensayos controlados, el 0.2% de los pacientes de sexo masculino se sometieron a la circuncisión. La mayoría de las infecciones micóticas genitales fueron tratadas con tratamientos antifungicos tópicos, prescritos por un profesional de la salud o por auto-tratamientos mientras continuaban el tratamiento con canagliflozina.
Consumo de alcohol: El alcohol es conocido por potenciar el efecto de la metformina en el metabolismo del lactato. Por lo tanto, se debe advertir a los pacientes contra el consumo excesivo de alcohol mientras están recibendo VOKANAMET™ (ver sección Advertencias y Precauciones).
Estados de hipoxia: El colapso cardiovascular (shock) por cualquier causa (por ejemplo, insuficiencia cardíaca congestiva aguda, infarto del miocardio agudo y otras condiciones caracterizadas por la hipoxemia) se ha asociado con acidosis láctica y también puede causar azotemia prerenal. Cuando ocurren tales eventos en pacientes con terapia con VOKANAMET™, el fármaco debe discontinuarse inmediatamente.
POSOLOGÍA Y ADMINISTRACIÓN:
Posología - Adultos de 18 años de edad o mayores: La dosis de VOKANAMET™ (canagliflozina y clorhidrato de metformina) debe individualizarse en base a la eficacia y la tolerabilidad, mientras no se exceda la dosis máxima recomendada de 150 mg de canagliflozina/1000 mg de clorhidrato de metformina dos veces al día. El aumento escalonado de la dosis debe ser gradual para reducir los efectos secundarios gastrointestinales (GI) asociados con el uso de la metformina.
Dosis de inicio recomendada:
• En pacientes actualmente tratados con canagliflozina y no tratados actualmente con metformina.
— En pacientes que están tomando canagliflozina 300 mg al día, cambiar a VOKANAMET™ 150 mg/850 mg dos veces al día con las comidas.
• En pacientes ya tratados con metformmina y no tratados con canagliflozina, cambiar a una dosis similar total diaria de metformina con 150 mg de canagliflozina tomada dos veces al día con los alimentos. Una dosis conteniendo 150 mg de canagliflozina dos veces al día puede considerarse para pacientes con una TFGe ≥ 60 mL/min/1.73 m2 [CrCl ≥ 60 mL/min], quienes necesitan un control glicémico estricto y quienes tienen un bajo riesgo de reacciones adversas relacionadas con el volumen intravascular reducido con el tratamiento de VOKANAMET™ (ver sección Poblaciones especiales a continuación y Advertencias y Precauciones).
En pacientes con evidencia de volumen intravascular reducido, se recomienda corregir esta condición antes de iniciar con VOKANAMET™ (Ver sección Advertencias y Precauciones).
• Pacientes ya tratados con componentes individuales de canagliflozina y metformina pueden ser cambiados a VOKANAMET™ conteniendo la misma dosis total diaria de cada componente que ya está siendo tomada o la dosis terapéuticamente adecuada más cercana.
En pacientes que tienen un TFGe ≥ 60 mL/min/1.73m2 [CrCl ≥ 60 mL/min] y quienes necesitan un control glucémico estricto, la dosis puede ser incrementada a VOKANAMET™ conteniendo 150 mg de canagliflozina tomada dos veces al día (ver sección Advertencias y Precauciones).
Cuando se utiliza VOKANAMET™ como complemento a la terapía con insulina o un secretagogo de insulina (por ejemplo, sulfonilurea), se puede considerar una dosis menor de insulina o secretagogo de insulina para reducir el riesgo de hipoglucemia (ver sección Advertencias y precauciones y Reacciones adversas).
ADMINISTRACIÓN: VOKANAMET™ debe tomarse por vía oral dos veces al día con los alimentos para reducir el riesgo de los efectos secundarios gastrointestinales asociados con el uso de metformina. Las tabletas deben ingerirse enteras.
Dosis omitida: Si se omite una dosis, ésta debe ser tomada tan pronto como el paciente lo recuerde a menos que sea casi la hora de la siguiente dosis, en cuyo caso, los pacientes deben omitir la dosis olvidada y tomar el medicamento en el momento de la siguiente dosis regular programada.
POBLACIONES ESPECIALES:
Población pediátrica (< 18 años de edad): No se ha establecido la seguridad y eficacia de VOKANAMET™ en pacientes pediátricos.
Ancianos: Debido al potencial de disminución de la función renal en sujetos ancianos, la dosis de VOKANAMET™ debe ajustarse en base a la función renal. Es necesaria la evaluación regular de la función renal (ver sección Advertencias y Precauciones).
Se debe tomar en consideración la función renal y el riesgo de reducción del volumen (ver sección Advertencias y Precauciones y Reacciones Adversas).
Insuficiencia renal: VOKANAMET™ no debe iniciarse en pacientes con una TFGe < 60 mL/min/1.73m2 [CrCl < 60 mL/min] y debe discontinuarse cuando la TFGe es persistentemente < 60 mL/min/1.73m2 [CrCl < 60 mL/min] (ver sección Advertencias y Precauciones).
SOBREDOSIS:
Canagliflozina: Las dosis únicas de hasta 1600 mg de canagliflozina en sujetos sanos y de canagliflozina 300 mg dos veces al día durante 12 semanas en pacientes con diabetes tipo 2 generalmente fueron bien toleradas.
Metformina: No se ha observado hipoglucemia con clorhidrato de metformina en dosis de hasta 85 g; aunque, ocurrió acidosis láctica en tales circunstancias. Altas sobredosis de metformina o riesgos concomitantes pueden conducir a acidosis láctica. La acidosis láctica es una emergencia médica y debe tratarse en el hospital.
Tratamiento: En caso de una sobredosis, es razonable emplear las medidas de soporte usuales, por ejemplo, remover el material no absorbido del tracto gastrointestinal, emplear monitorización clínica e instaurar tratamiento de soporte según el estado clínico del paciente. El método más efectivo para remover el lactato y la metformina es la hemodiálisis. La eliminación de la canagliflozina durante una sesión de hemodiálisis de 4 horas fue insignificante. No se espera que canagliflozina sea dializable por diálisis peritoneal.
PRESENTACIÓN:
Naturaleza y contenido del envase: Frasco a prueba de niños HDPE, sellado por inducción y desecante.
Tamaño de los frascos de 20 y 60 tabletas recubiertos.
Instrucciones de uso y manipulación: No hay requisitos especiales.
Instrucciones para la eliminación: Cualquier producto no utilizado o material de desecho debe eliminarse de acuerdo con las normas locales.
Presentación:
VOKANAMET™ Tabletas recubiertas 150 mg/850 mg (Reg. San. INVIMA 2015M-0016489).
VOKANAMET™ Tabletas recubiertas 150 mg/1000 mg (Reg. San. INVIMA 2015M-0016488).
JANSSEN-CILAG, S. A.
Av. Calle 26 No. 69-76 - Edificio Elemento
Torre 2, Piso 11 - PBX: +57 1 927 1200 - Bogotá, D. C.
CONDICIONES DE ALMACENAMIENTO: Observe las precauciones de conservación en el embalaje exterior. Mantenga fuera de la vista y del alcance de los niños.


