

XYNTHA
MOROCTOCOG ALFA
Polvo liofilizado para reconstituir
Caja , 1 Vial(es) , Polvo , 250 y 500 Unidades Internacionales
Caja , 1 Vial(es) , Polvo , 1000 y 200 Unidades Internacionales
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN Y CARACTERÍSTICAS FARMACÉUTICAS: Viales de único uso que nominalmente contienen 250, 500, 1000 o 2000 Unidades Internacionales (UI) de moroctocog alfa (factor VIII de la coagulación recombinante) por vial.
Excipientes: Polisorbato 80 (0,4 mg/vial), Sacarosa (12 mg/vial), L-Histidina (6 mg/vial), Cloruro de Calcio Dihidratado (1 mg/vial), Cloruro de Sodio (72 mg/vial después de reconstituido con el diluyente).
INDICACIONES: XYNTHA® está indicado para el control y prevención de episodios hemorrágicos y para la profilaxis de rutina y quirúrgica en pacientes con hemofilia A (deficiencia congénita del factor VIII o hemofilia clásica).
XYNTHA® no contiene el factor von Willebrand y por lo tanto no está indicado para la enfermedad de Von Willebrand.
MODO DE ACCIÓN: El factor VIII activado actúa como un cofactor para el factor IX activado que acelera la conversión del factor X a factor X activado. El factor X activado convierte protrombina en trombina. La trombina convierte fibrinógeno en fibrina y se forma un coágulo. La actividad del factor VIII es muy reducida en pacientes con hemofilia A y por ello es necesaria la terapia de reemplazo. La administración de XYNTHA® aumenta los niveles plasmáticos de la actividad del factor VIII y pueden corregir temporalmente el problema de coagulación de estos pacientes.
USO GERIÁTRICO: Los estudios clínicos de XYNTHA® no incluyeron sujetos de 65 o más años de edad. En general, se debe individualizar la selección de la dosis para los pacientes ancianos.
USO PEDIÁTRICO: Se estudió la seguridad de XYNTHA® en niños y adolescentes previamente tratados (n=18, 12-16 años de edad en el estudio principal y n=49, 7-16 años de edad en el estudio de respaldo). En el estudio principal, los datos sobre eventos adversos de pacientes con = 16 años de edad se compararon con los datos de pacientes de más 16 años de edad. Dieciocho (18) pacientes = 16 años de edad y 76 de >16 años de edad. El nivel de exposición en ambos grupos de pacientes fue similar. Los eventos adversos que surgieron durante el tratamiento fueron similares en severidad e incidencia en los dos grupos de edad.32 Ensayos clínicos que evalúan la utilización de moroctocog alfa (AF-CC) en PUP (estudio 4434), con niños menores de 6 años de edad (estudio 313) y niños menores de 12 años de edad (estudio 4433) se encuentran actualmente en curso.
XYNTHA® puede utilizarse de la misma forma que se utilizaba el producto predecesor ReFacto, debido a que son bioquímicamente comparables y ha demostrado características farmacocinéticas similares con el producto predecesor ReFacto. Se ha estudiado la seguridad y eficacia del producto predecesor ReFacto en niños y adolescentes previamente tratados (n = 31, 5-18 años de edad) y en neonatos, infantes y niños no tratados previamente (n=101, <1-52 meses de edad). Ver Farmacocinética.
COMPATIBILIDADES, INCOMPATIBILIDADES: Debido a la ausencia de estudios de incompatibilidad, no se debe administrar XYNTHA® reconstituido en el mismo tubo o contenedor con otros medicamentos. Los componentes del kit para administración de infusión suministrados en esta caja son compatibles con XYNTHA®.
CLASE FARMACOLÓGICA, CLASE TERAPÉUTICA: Antihemorrágicos: Factor VIII de la coagulación sanguínea.
Código ATC: B02BD02.
FARMACOCINÉTICA: En un estudio clínico transversal principal, (estudio 310) XYNTHA® demostró equivalencia farmacocinética con Advate®,* otro producto del factor VIII recombinante, en 30 pacientes previamente tratados (? 12 años) utilizando el método de bioequivalencia estándar basado en el ensayo de la coagulación de una fase. Los cocientes de las medias geométricas de mínimos cuadrados de XYNTHA® y Advate® fueron 100%, 89,8% y 88,0% para los valores K, AUCt y AUC?, respectivamente. Los intervalos de confianza del 90% correspondientes sobre los cocientes de las medias geométricas de XYNTHA® y Advate® se encontraron dentro de la ventana de equivalencia de 80% a 125%, lo que demuestra equivalencia farmacocinética de XYNTHA® y Advate®.
(*Advate® es una marca comercial registrada de Baxter International, Inc.)
En el mismo estudio, los parámetros farmacocinéticos de XYNTHA® fueron determinados al inicio del estudio y durante el seguimiento para 25 pacientes tratados previamente (=12 años) después de administración repetida de XYNTHA® durante seis meses. Luego de una única infusión intravenosa de 2 minutos de una dosis de 50 UI/kg de XYNTHA®, el FVIII:C plasmático aumentó abruptamente con una Cmax media (±DE) de 1,12 (?0,19) UI/mL. , De ahí en adelante, la disminución del FVIII:C mostró una disposición bifásica característica. En la fase inicial, la actividad cayó a una tasa consistente con una distribución en el espacio intravascular relativamente rápida, pero limitada. La media (±DE) del volumen de distribución en estado estable fue de 65,1 (?35,1) mL/kg. Durante la fase terminal, la tasa de disminución del FVIII:C fue más lenta que en la fase inicial con una vida media de eliminación terminal media (?DE) de 11,8 (?5,1) horas. Un perfil farmacocinético similar se obtuvo después de repetir la utilización durante seis meses (ver Tabla 2). Los cocientes de los mínimos cuadrados medios geométricos de la farmacocinética a los 6 meses y la farmacocinética al inicio del estudio fueron 107%, 100% y 104% para el valor K, AUCt y AUC?, respectivamente. Los intervalos de confianza del 90% correspondientes a los cocientes entre el mes 6 y el inicio del estudio para los parámetros farmacocinéticos anteriores se encontraron dentro de la ventana de bioequivalencia de 80% y 125%. Esto indica que no se presentó ningún cambio dependiente del tiempo en las propiedades farmacocinéticas de XYNTHA®.
|
Tabla 2. |
|||||
|
Estimados de los Parámetros Farmacocinéticos para XYNTHA® al Inicio del Estudio y en el Mes 6 en Pacientes con Hemofilia A Previamente Tratados |
|||||
|
Consulta |
Cmax (UI/mL) |
AUCt |
AUC? |
Valor K |
Recuperación |
|
Inicio del estudio |
|||||
|
Media ? de (Mín., Máx.) |
1,12 ? 0,19 |
13,3 ? 5,2 |
14,2 ? 5,5 |
2,23 ? 0,39 |
105 ? 19 |
|
(0,59; 1,41) |
(4,1; 23,6) |
(4,7; 25,0) |
(1,19; 2,83) |
(53,4; 132) |
|
|
n |
25 |
25 |
25 |
25 |
25 |
|
Mes 6 |
|||||
|
Media ? DE |
1,24 ? 0,42 |
13,3 ? 6,7 |
15,0 ? 7,5 |
2,47 ? 0,84 |
116 ? 40 |
|
(Mín., Máx.) |
(0,65; 2,60) |
(5,0; 41,0) |
(5,3; 42,0) |
(1,29; 5,20) |
(59,3; 256) |
|
n |
25 |
25 |
25 |
25 |
25 |
|
Abreviaturas: AUC? = área bajo la curva de concentración plasmática-tiempo desde el tiempo cero hasta el infinito; AUCt = área bajo la curva de concentración plasmática–tiempo desde cero hasta la última concentración medible; Cmax = concentración máxima; valor K = recuperación incremental; DE=desviación estándar |
|||||
En un estudio principal de fase III (estudio 311) de profilaxis quirúrgica, se evaluó la farmacocinética de XYNTHA® durante el manejo perioperatorio de pacientes con hemofilia A que se sometieron a cirugía mayor. En la consulta al inicio del estudio, todos los pacientes recibieron una única dosis de XYNTHA® de aproximadamente 50 UI/kg. Se analizaron las muestras para la actividad de FVIII utilizando el método de coagulación validado de única fase (UE). Se encontraban disponibles los datos de recuperación para un total de 30 pacientes; el valor K medio (? desviación estándar [DE]) fue 2,11 (? 0,43) UI/dL por UI/kg, y el valor de la recuperación in vivo media (±DE) fue 101.0% (? 20%).
En pacientes no tratados previamente (PNP), los parámetros farmacocinéticos del producto predecesor ReFacto se evaluaron utilizando el ensayo cromogénico. Estos pacientes (n=59; media de la edad 10 ? 8,3 meses) presentaron una recuperación incremental media a la Semana 0 de 1,5 ? 0,6 UI/dL por UI/kg (intervalo 0,2-2,8 UI/dL por UI/kg), que fue menor que la obtenida en los PTP a la Semana 0 con un valor K medio de 2,4 + 0,4 UI/dL por UI/kg (intervalo 1,1-3,8 UI/dL por UI/kg). La recuperación incremental media fue estable en el tiempo (5 consultas durante un periodo de 2 años) y varió entre 1,5 y 1,8 UI/dL por UI/kg. El modelo farmacocinético de la población utilizando los datos de 44 PNP produjo un valor medio de la semivida estimada de 8.0 ? 2,2 horas.
FARMACODINÁMICA, EFICACIA CLÍNICA: XYNTHA®, factor VIII de la coagulación recombinante es una glucoproteína con una masa molecular aproximada de 170.000 Da, se compone de 1.438 aminoácidos, que no contienen el dominio B no funcional. , XYNTHA® es una sustancia basada en ADN recombinante que tiene características funcionales similares al del factor VIII endógeno.
El complejo de factor VIII/factor de von Willebrand se compone de dos moléculas, (el factor VIII y el factor von Willebrand) con funciones fisiológicas diferentes. Cuando se aplica por infusión en un paciente hemofílico, el factor VIII se une al factor von Willebrand en la circulación del paciente.
El factor VIII activado actúa como cofactor del factor IX activado acelerando la conversión de factor X a factor X activado. El factor X activado convierte protrombina en trombina. La trombina convierte el fibrinógeno en fibrina y se forma un coágulo.
La hemofilia A es un trastorno de la coagulación sanguínea hereditario vinculado al cromosoma X debido a la disminución de los niveles del factor VIII:C y produce hemorragias en las articulaciones, los músculos o los órganos internos, de forma espontánea o como resultado de un trauma accidental o quirúrgico. Mediante la terapia de reemplazo, los niveles plasmáticos del factor VIII se aumenta, permitiendo de esta forma una corrección temporal de la eficiencia del factor y una corrección de la tendencia de hemorragia.
En un estudio principal de fase 3, se evaluó la eficacia de XYNTHA® para profilaxis rutinaria y tratamiento por demanda. La profilaxis se inició a una dosis de 30 UI/kg administradas 3 veces por semana. El régimen posológico en el tratamiento por demanda fue determinado por el investigador. Noventa y cuatro (94) PTP con hemofilia A moderadamente grave a severa (FVIII:C ?2%) recibieron al menos una dosis de XYNTHA® y fueron incluidos en la población con intención de tratar (ITT). En este estudio 89 pacientes acumularon al menos 50 días de exposición (DE) con XYNTHA®.
De los 94 pacientes de la población ITT, 30 pacientes con FVIII:C ?1% participaron también en el periodo PK transversal, doble ciego, aleatorio de estudio y fueron incluidos en la población por protocolo para análisis de la equivalencia farmacocinética con relación a otro producto FVIIIr, Advate® y para caracterización completa PK. Los resultados de estos análisis muestran que XYNTHA® es farmacocinéticamente equivalente a Advate®, y el perfil farmacocinético de XYNTHA® permaneció estable después de 6 meses de utilización continua. ,
El análisis por intensión de tratamiento de las variables de eficacia clínica en el periodo de análisis de la seguridad y eficacia de etiqueta abierta produjo resultados positivos similares. Todos los 94 pacientes recibieron XYNTHA® para profilaxis de rutina; la dosis media administrada fue 30,2 UI/kg (intervalo, 6,8 a 76,9 UI/kg). La mayoría de los pacientes (57/94; 60,6%) no reportaron hemorragias espontáneas mientras recibían profilaxis de rutina. La tasa media de hemorragia anualizada (TMH) para todos los episodios de hemorragia fue 1,9 (media 3,9, intervalo 0 a 42,1), que indica prevención efectiva de la hemorragia en la población del estudio. Cincuenta y tres (53) de los 94 pacientes recibieron XYNTHA® durante el tratamiento por demanda. La dosis media administrada fue 30,6 UI/kg (intervalo, 6,4 a 74,4 UI/kg). La mayoría de los episodios de hemorragia (173/187; 92,5%) se resolvieron con 1 o 2 infusiones. Estos resultados no se limitaron a ubicaciones específicas de hemorragia, ya que la eficacia fue similar en las hemorragias que ocurrían en las articulaciones, los tejidos suaves/los músculos y otros sitios. Se utilizó un rango amplio de dosis para iniciar el tratamiento de la hemorragia; sin embargo, la distribución de las dosis utilizadas para iniciar el tratamiento de la hemorragia fue similar sin importar la ubicación de la hemorragia. Los pacientes calificaron la mayoría de las infusiones utilizadas para iniciar el tratamiento de la hemorragia como excelentes o buenas (132/187; 70,6%). La incidencia del efecto terapéutico menor al esperado (LETE) ocurrió a una tasa de 0,4% (25/6.404 infusiones profilácticas) cuando se administró XYNTHA® para profilaxis y 0,5% (1/187 episodios de hemorragia) cuando se administró para tratamiento por demanda.
Un estudio principal de fase 3 (estudio 311) para profilaxis quirúrgica en pacientes con hemofilia A incluyó PTP con hemofilia A severa o moderadamente severa (FVIII:C=2%) sometidos a procedimientos quirúrgicos mayores que recibieron XYNTHA® o ReFacto AF. Treinta (30) pacientes fueron tratados con XYNTHA® o ReFacto AF y conformaron la población ITT; 29 pacientes fueron sometidos a cirugía mayor y completaron el estudio. Treinta pacientes (30) fueron asignados para recibir XYNTHA® por inyección de bolo (IB; 22 pacientes) o por infusión continua (IC; 8 pacientes) a criterio del médico para apoyar la hemostasia quirúrgica luego de atención hospitalaria y atención ambulatoria posoperatorias. Un sujeto asignado a IC recibió XYNTHA® únicamente durante una evaluación farmacocinética prequirúrgica y no fue sometido a cirugía. Los 22 pacientes tratados con IB recibieron un total de 942 infusiones (que varían entre 16 y 72 infusiones por paciente) para un total acumulado de dosis de 2.037.386 UI de XYNTHA® durante 682 días de exposición (DE) total acumulada (que variaron entre 15 y 40 DE por paciente). Los 8 pacientes asignados al tratamiento con IC, incluyendo un paciente que recibió solamente una dosis para evaluación PK, recibieron una dosis total de 529.977 UI de XYNTHA® durante 204 DE en total (intervalo 1 a 37 DE por paciente).
De los 29 pacientes que fueron sometidos a cirugía, 25 fueron incluidos en la población evaluable para eficacia. Los procedimientos quirúrgicos mayores para los 25 pacientes evaluables para eficacia fueron 11 artroplastias totales de rodilla, 1 artroplastia de cadera, 5 sinovectomías, 1 liberación de transposición del nervio cubital, 1 reparación de hernia ventral/revisión cicatricial, 1 artroscopia de rodilla, 1 revisión y desbridamiento de rodilla después de artroscopia total de rodilla, 1 revisión de artroplastia de cadera, 1 estapedotomía, 1 artrodesis de tobillo y 1 extirpación de pseudotumor. Para los 25 pacientes quirúrgicos las calificaciones del investigador, al final de la cirugía y al final del periodo postoperatorio inicial fueron excelentes o buenas para todas las evaluaciones, la pérdida de sangre intraoperatoria fue reportada como normal o ausente para todos los procedimientos. Trece de los 25 pacientes evaluables presentaron pérdida de sangre en el periodo postoperatorio y en 10 casos la pérdida de sangre postoperatoria fue calificada normal. En 3 casos la pérdida postoperatoria de sangre fue calificada anormal: 1 debido a hemorragia después de trauma quirúrgico de la arteria epigástrica, 1 debido a una pérdida de sangre de 800 mL después de cirugía de artroplastia de cadera y 1 después de sinovectomía del codo en la que la pérdida de sangre no pudo medirla el investigador.
En un estudio previo de respaldo (estudio 306) de XYNTHA®, se clasificaron 6 procedimientos quirúrgicos como mayores según las definiciones del estudio quirúrgico principal. En todos los casos, la eficacia hemostática fue manejada efectivamente con XYNTHA®. Ningún paciente presentó pérdida de sangre mayor a 50 mL, y no se realizó ninguna transfusión sanguínea.
CONTRAINDICACIONES: XYNTHA® puede estar contraindicado en pacientes con hipersensibilidad a alguno de los componentes de la preparación.
XYNTHA® no se ha estudiado en pacientes con antecedentes de hipersensibilidad a las proteínas de hámster. XYNTHA® puede estar contraindicado en pacientes con antecedentes de hipersensibilidad a alguno de los componentes de la preparación y en pacientes con antecedentes de hipersensibilidad a proteínas de hámster.
EMBARAZO: No se han realizado estudios de reproducción animal con XYNTHA®. Debido a la ocurrencia poco frecuente de hemofilia A en mujeres, no se encuentra disponible experiencia relacionada con la utilización del factor VIII durante el embarazo. Por tanto, los productos del factor VIII se deben administrar a mujeres embarazadas únicamente si está claramente indicado.
LACTANCIA: No se han realizado con XYNTHA® estudios de reproducción animal. Se desconoce si el medicamento se elimina en la leche materna. Debido a la ocurrencia poco frecuente de hemofilia A en mujeres, no se encuentra disponible experiencia relacionada con la utilización de productos del factor VIII durante la lactancia. Por lo tanto, los productos del factor VIII debe administrarse a mujeres que están lactando únicamente si está claramente indicado.
EFECTOS SOBRE LAS ACTIVIDADES QUE REQUIEREN CONCENTRACIÓN Y DESEMPEÑO: No se han realizado estudios sobre efectos en la habilidad para manejar y utilizar máquinas.
REACCIONES ADVERSAS: En las siguientes tablas, se utilizan las categorías de frecuencia y los términos de CIOMS:
Muy común ?10%
Común ?1%
Poco común ?0,1% y ?1%
Raro ?0,01% y ?0,1%
Muy raro <0,01%
|
Clase de sistema de órganos |
|||||
|
(Trastorno) |
Muy frecuente |
Frecuente |
Poco frecuente |
Raro |
Muy raro |
|
Trastornos del Sistema inmunitario |
Reacción anafilactoide |
||||
|
Trastornos Cardíacos |
Angina de pecho, taquicardia, palpitaciones* |
||||
|
Investigaciones |
Aumento en exámenes de laboratorio de anticuerpos para IgG de ratón (únicamente para ReFacto). Aumento en exámenes de laboratorio de anticuerpos para FVIII. Aumento en exámenes de laboratorio de anticuerpos para proteína de ovarios de hámster chino (CHO) |
Aumento de CPK, Aumento de aspartato aminotransferasa. Aumento de alanina aminotransferasa*, Aumento de bilirrubina |
|||
|
Trastornos del Sistema Nervioso |
Cefalea |
Mareo, Neuropatía*, Aumento de la transpiración, somnolencia, disgeusia |
|||
|
Trastornos del metabolismo y la nutrición |
Anorexia |
||||
|
Trastornos musculoesqueléticos y del tejido conectivo |
Artralgia |
Mialgia |
|||
|
Trastornos vasculares |
Hemorragia/hematoma, Rubefacción*, Tromboflebitis*, Hipotensión, Vasodilación |
||||
|
Trastornos respiratorios, torácicos y mediastínicos |
Aumento de la tos, Disnea |
||||
|
Trastornos Gastrointestinales |
Vómito*, Náusea |
Diarrea |
|||
|
Trastornos de la piel y el tejido subcutáneo |
Prurito |
Exantema, Urticaria |
|||
|
Trastornos Generales y condiciones en el sitio de administración |
Fiebre, Astenia, Escalofríos, Complicaciones del catéter de acceso venoso permanente, dolor en el sitio de Inyección, reacción en el sitio de Inyección, inflamación en el sitio de Inyección* |
||||
|
Inhibición del Factor VIII† |
Inhibición del factor VIII en Pacientes No Tratados Previamente (PNP) |
Inhibición del factor VIII en Pacientes Tratados Previamente(PTP) |
|||
|
Las frecuencias de las reacciones adversas se calculan como eventos por infusión |
|||||
|
(*) = Estas reacciones adversas fueron totalizadas a partir de eventos adversos y eventos de hemofilia en todos los estudios sin importar su relación con el medicamento del estudio. Todas las demás reacciones adversas fueron totalizadas para todos los estudios a partir de eventos adversos relacionados con el medicamento bajo estudio y los eventos de hemofilia ÚNICAMENTE Para las frecuencias de las reacciones adversas, en los pacientes quirúrgicos que estaban recibiendo infusión continua (IC), cualquier vía de administración de IC se considera una infusión. (†) = La frecuencia de la Reacción Adversa, inhibición del Factor VIII, se expresa por paciente. |
|||||
La reacción adversa debida al tratamiento por infusión, reportada con mayor frecuencia, fue el vómito . La mayoría de las reacciones reportadas fueron consideradas de severidad leve o moderada.
Además, como ocurre con todos los productos intravenosos de proteínas, son posibles reacciones de hipersensibilidad del tipo alérgicas. Las manifestaciones de reacciones de hipersensibilidad pueden incluir urticaria, urticaria generalizada, presión en el pecho, sibilancias, hipotensión y anafilaxia.
Si ocurre alguna reacción que se considera relacionada con la administración de Moroctocog Alfa (AF-CC), la tasa de infusión debe disminuirse o interrumpir la infusión, como lo indique la respuesta del paciente.
Los pacientes con hemofilia A pueden desarrollar anticuerpos neutralizantes (inhibidores) para el Factor VIII. Ver también Inhibidores y “PRECAUCIONES”. Como ocurre con todos los productos del factor de coagulación VIII, debe controlarse a los pacientes con relación al desarrollo de inhibidores que son cualificados en Unidades Bethesda (UB) utilizando la modificación de Nijmegen del ensayo Bethesda. Si dichos inhibidores ocurren, la condición puede manifestarse por sí misma como una respuesta clínica insuficiente o un rendimiento bajo inesperado de la actividad plasmática del Factor VIII. En tales casos, se recomienda contactar un centro especializado en hemofilia.
El riesgo de desarrollar inhibidores se correlaciona con la exposición al factor VIII antihemofílico, este riesgo es el más alto durante los primeros 20 días de exposición. Raramente, pueden desarrollarse inhibidores después de los primeros 100 días de exposición.
Se han observado casos de inhibidor recurrente (titulación baja) después de cambiar desde un producto de FVIII a otro en pacientes previamente tratados con más de 100 días de exposición que tienen antecedentes previos de desarrollo de inhibidores. Por lo tanto, se recomienda controlar cuidadosamente a los pacientes con relación a la ocurrencia de inhibidores cuando se realice cambio a otro producto.
Se han recibido informes de falta de efecto, principalmente en pacientes bajo profilaxis durante ensayos clínicos y en entornos postcomercialización. La falta de efecto y/o la baja recuperación del factor VIII se han reportado en pacientes con inhibidores pero también en pacientes que no presentan ninguna evidencia de inhibidores. La falta de efecto se ha descrito como hemorragia en las articulaciones objetivo, hemorragia en nuevas articulaciones, otras hemorragias o sensaciones subjetivas del paciente de inicio de nuevas hemorragias. Para garantizar una respuesta terapéutica adecuada es importante, TITULAR Y CONTROLAR INDIVIDUALMENTE la dosis de XYNTHA® de cada paciente, especialmente cuando se inicia el tratamiento con XYNTHA®.30
En un estudio de fase 3 principal (estudio 310), en el que pacientes previamente tratados (PPT) con hemofilia A recibieron XYNTHA® para profilaxis de rutina y tratamiento por demanda, 94 pacientes recibieron al menos una dosis de XYNTHA® que resultó en un total de 6775 infusiones. En este estudio, la incidencia de los inhibidores del FVIII para XYNTHA® fue el criterio de valoración de la seguridad principal. En dos pacientes con titulación baja, de estos 94 pacientes (2,1%), se observaron inhibidores transitorios.
En un estudio complementario de XYNTHA® (estudio 306), fueron observados 1 inhibidor de novo y dos inhibidores recurrentes (todos con titulación baja determinada por el laboratorio central) en 110 pacientes; la mediana de la exposición fue de 58 días de exposición (DE) (intervalo 5-140) y 98 pacientes tuvieron al menos 50 DE a XYNTHA®. Noventa y ocho (98) de los 110 pacientes originales continuaron el tratamiento en un segundo estudio complementario (307) y extendieron la exposición a XYNTHA® con una mediana de 169 DE adicionales (intervalo 9-425). Se observó un (1) inhibidor adicional de novo de baja titulación. La frecuencia de inhibidores observada en estos estudios se encuentra dentro del intervalo esperado. , ,
En un ensayo clínico de XYNTHA® fabricado mediante el proceso anterior (estudio 300), uno de los 113 pacientes (0,9%) sometidos previamente a múltiples tratamientos que fueron evaluados para eficacia en episodios hemorrágicos desarrollaron inhibidor de alta titulación. El desarrollo de inhibidor en este paciente ocurrió en el mismo lapso de tiempo que el desarrollo de gammapatia monoclonal de importancia incierta. El paciente fue inicialmente observado en un laboratorio local presentando inhibidor de baja titulación debida al tratamiento a los 98 días de exposición lo que se confirmó a 2 UB en el laboratorio central a los 113 días de exposición. Después de 18 meses bajo tratamiento con XYNTHA®, la concentración del inhibidor aumentó a casi 13 UB y un episodio de hemorragia no respondió al tratamiento con XYNTHA®.
En un análisis estadístico Bayesiano, los resultados del estudio 310 (dos de 94 pacientes desarrollaron un inhibidor, 89 presentaron 50 o más días de exposición a XYNTHA®) se utilizaron para actualizar los resultados de los PPT de estudios complementarios previos de XYNTHA®, en los que se observó un inhibidor de novo y dos inhibidores recurrentes en 110 pacientes y un inhibidor fue observado en 113 pacientes. Este análisis Bayesiano indica que la tasa de población con inhibidor (verdadero) para XYNTHA® estuvo bajo un valor aceptable preestablecido del 4,4%; el estimado del límite superior del 95% de la tasa de inhibidor verdadero fue de 4,07%.
En un estudio principal de fase 3 de profilaxis quirúrgica en pacientes con hemofilia A (estudio 311), un inhibidor persistente de baja titulación y un inhibidor falso-positivo transitorio fueron reportados.
En un ensayo clínico (estudio 301), 32 de 101 pacientes (32%) previamente no tratados sometidos a tratamiento con XYNTHA® [fabricado por el proceso anterior] desarrollaron inhibidores: 16 de 101 (16%) con una titulación >5 Unidades Bethesda (UB) y 16 de 101 (16%) con una titulación =5 UB.23 La mediana de los días de exposición antes de desarrollar inhibidores en estos pacientes fue 12 días (intervalo 3 - 49 días). De los 16 pacientes que presentaron alta respuesta al tratamiento, 15 recibieron tratamiento de tolerancia inmunitaria (TI). Once (11) de los que presentaron respuesta alta al tratamiento tuvieron una titulación <0,6 UB en su más reciente prueba disponible después del TI. Además, el tratamiento TI se inició en 10 de los 16 pacientes con baja titulación (=5 UB), 9 de los cuales presentaron titulación <0,6 UB para su valor más reciente. Por lo tanto, el TI mostró una eficacia general del 80% (20/25), 73% para los que presentaron alta respuesta el tratamiento y 90% para los que presentaron baja respuesta al tratamiento. En cinco (5) de los 6 pacientes restantes que presentaron baja respuesta y que no recibieron TI también se observó una titulación <0,6 UB para su más reciente valor.22
Han existido informes postcomercialización espontáneos de desarrollo de inhibidores de alta titulación en pacientes previamente tratados.28
Se han observado en ensayos clínicos aumentos en el laboratorio de las titulaciones de anticuerpos anti-FVIII, sin desarrollo de inhibidores. En un estudio de PTP que estaban recibiendo XYNTHA® para tratamiento de rutina y prevención de episodios hemorrágicos (estudio 310) y para profilaxis quirúrgica (estudio 311), 1 de 94 pacientes (1%), y 1 de 30 pacientes (3%), respectivamente, desarrollaron anticuerpos anti-FVIII; estos pacientes no desarrollaron un inhibidor. La significancia clínica de estos anticuerpos, en ausencia de un inhibidor no es clara.
En ensayos clínicos de PTP que estaban recibiendo XYNTHA® para tratamiento y prevención de rutina de episodios hemorrágicos, 0 de 94 pacientes (0%) en el estudio 310 y 3 de 110 pacientes (3%) en el estudio 306/307, desarrollaron un aumento en el laboratorio en la titulación de anticuerpos anti-CHO (ovarios de hámster chino, la estirpe celular que es la fuente del factor VIII para XYNTHA®), sin ningún efecto clínico aparente. En un estudio con XYNTHA® para profilaxis quirúrgica (estudio 311) 1 de 30 pacientes (3%) desarrolló un aumento en el laboratorio de anticuerpos para CHO. Veinte (20) de 113 (18%) PTP que estaban recibiendo XYNTHA® fabricado mediante el proceso anterior (estudio 300) presentaron aumento en la titulación de anticuerpos anti-CHO, sin ningún efecto clínico aparente.
La seguridad de XYNTHA® fue evaluada en niños y adolescentes previamente tratados (n=18, edad en un estudio [estudio 310]; y n=49, edad en un estudio complementario [estudio 306]). , Aunque se ha estudiado un número limitado de niños, existe una tendencia de mayores frecuencias de eventos adversos en niños de 7-16 años de edad cuando se comparan con los adultos.
No existe ningún dato clínico de pacientes no tratados previamente (PNP) que se sometieron a tratamiento con XYNTHA®. Los ensayos clínicos que evalúan la utilización de moroctocog alfa (AF-CC) en PNP (estudio 4434), en niños menores de 6 años de edad (estudio 313) y niños menores de 12 años de edad (estudio 4433) están en curso.
INTERACCIONES: No se conocen interacciones de los productos del factor VIII de la coagulación recombinante con otros medicamentos.
INTERACCIONES CON PRUEBAS DE LABORATORIO Y OTRAS PRUEBAS DE DIAGNÓSTICO: No aplica
DATOS DE SEGURIDAD PRECLÍNICA: No se han realizado estudios con XYNTHA® para evaluar su potencial mutágeno o carcinógeno. XYNTHA® ha demostrado similitud con el producto predecesor ReFacto con relación a sus propiedades bioquímicas y fisicoquímicas, y en su farmacología y toxicología in vivo no clínica. Por inferencia se podría esperar que el producto predecesor ReFacto y XYNTHA® tengan potencial mutágeno y carcinógeno equivalentes. El producto predecesor ReFacto demostró en el ensayo de micronúcleos de ratón que no es genotóxico. No se han realizado estudios en animales para evaluar la carcinogenia, el deterioro de la fertilidad o del desarrollo fetal.
En estudios preclínicos, XYNTHA® se utilizó para restauración segura y efectiva de la hemostasia. XYNTHA® demostró un perfil toxicológico similar al perfil toxicológico observado en el producto predecesor ReFacto, que a su vez ha demostrado perfil toxicológico similar a un producto del factor VIII derivado del plasma cuando se someten a pruebas de dosis repetidas en estudios toxicológicos con animales.
PRECAUCIONES: En los pacientes que reciben productos que contienen el factor VIII de la coagulación se pueden desarrollar anticuerpos neutralizantes de su actividad (inhibidores). Como sucede con todos los productos que contienen el factor VIII de la coagulación, se debe controlar a los pacientes para determinar si se presenta el desarrollo de inhibidores que deben ser titulados en Unidades Bethesda utilizando las pruebas biológicas apropiadas. Si no se obtienen los niveles de actividad plasmática esperados del factor VIII o si no se controla la hemorragia con una dosis apropiada, se deberá realizar un ensayo para determinar si se encuentra presente un inhibidor del factor VIII.
Estos inhibidores usualmente son inmunoglobulinas IgG que se dirigen contra la actividad procoagulante del factor VIII que se identifican en UB utilizando el ensayo Bethesda. El riesgo de desarrollar inhibidores se correlaciona con la exposición al factor VIII antihemofílico, este riesgo es mayor dentro de los primeros 20 días de exposición. Los inhibidores son comunes en pacientes no tratados previamente , , y se han observado en pacientes tratados previamente con productos del factor VIII.
Se recomienda que, cuando sea posible, todas las veces que XYNTHA® se administre a los pacientes, se documente el nombre y número de lote del producto.
ABUSO Y DEPENDENCIA: El Factor Antihemofílico (Recombinante), Libre de Plasma/Albúmina no tiene potencial para abuso. No existe evidencia de dependencia del Factor Antihemofílico (Recombinante), Libre de Plasma/Albúmina.
ADVERTENCIAS ESPECIALES: Como ocurre con todos los productos de proteínas administrados vía intravenosa, se pueden presentar reacciones de hipersensibilidad del tipo alérgico. Debe informarse a los pacientes de los signos iniciales de hipersensibilidad (que incluyen ronchas, urticaria generalizada, opresión en el pecho, jadeo e hipotensión) y anafilaxia.
Si se presentan reacciones alérgicas o de anafilaxia, debe interrumpirse inmediatamente la administración de XYNTHA® y proporcionar manejo médico apropiado, que puede incluir el tratamiento para choque. Si alguno de los síntomas descritos ocurre, debe aconsejarse a los pacientes para que descontinúen el medicamento y contacten al médico y/o a un centro de urgencias dependiendo del tipo y la severidad de la reacción.
POSOLOGÍA Y ADMINISTRACIÓN: El tratamiento con XYNTHA® debe iniciarse bajo supervisión de un médico con experiencia en el tratamiento de la hemofilia A.
La posología y duración del tratamiento dependerá de la gravedad de la deficiencia del factor VIII, el lugar y nivel de hemorragia y la condición clínica del paciente. , La respuesta al factor VIII puede variar en cada paciente, lográndose niveles diferentes de recuperación in vivo y demostrando diferentes vidas medias. Las dosis administradas se deben titular de acuerdo con la respuesta clínica del paciente. , En presencia de un inhibidor, es posible que se necesiten dosis mayores o tratamientos específicos apropiados. No se ha evaluado en estudios clínicos el ajuste de la dosis en pacientes con insuficiencia renal o deterioro hepático.
XYNTHA® puede utilizarse en adultos y niños. Ver también Uso pediátrico.
El número de unidades de factor VIII administradas se expresa en Unidades Internacionales (UI), que corresponden con la norma vigente de la OMS para productos del factor VIII. La actividad plasmática del factor VIII se expresa como un porcentaje (con relación al plasma humano normal) o en Unidades Internacionales (con relación al Estándar Internacional para el factor VIII en el plasma).
Una Unidad Internacional (UI) de la actividad del factor VIII corresponde aproximadamente a la cantidad de factor VIII en un ml de plasma humano normal. El cálculo de la dosis requerida de factor VIII se basa en el hallazgo empírico de que, en promedio, una (1) UI de factor VIII por kg de peso corporal aumenta la actividad plasmática del factor VIII en 2 UI/dL. La dosis requerida se determina utilizando la siguiente fórmula:
Unidades requeridas = peso corporal (kg) x aumento deseado del factor VIII (% del valor normal o UI/dL) x 0,5 (UI/kg por UI/dL)
La potencia especificada en la etiqueta de XYNTHA® se basa en el ensayo de sustrato cromogénico de la Farmacopea Europea que se utiliza para calibrar el estándar de potencia en el proceso de fabricación de Wyeth utilizando un ensayo de coagulación de una fase. Este método de asignación de potencia está diseñado para armonizar XYNTHA® con el control clínico que utiliza un ensayo de coagulación de una fase. Con los productos del factor VIII recombinante, los controles clínicos que utilizan el ensayo cromogénico normalmente obtienen resultados que son mayores a los resultados obtenidos con el ensayo de coagulación de una fase.
Los datos clínicos respaldan la utilización del ensayo de coagulación de una fase para controlar la terapia con XYNTHA®.
Con base en su régimen de tratamiento actual, deberá recomendarse a las personas con hemofilia A que, cuando realicen un viaje, lleven el suministro adecuado del producto del factor VIII para los tratamientos necesarios. Deberá recomendarse a los pacientes consultar con su médico antes de viajar.
Debe considerarse el control preciso de la terapia sustitutiva utilizando un ensayo de la actividad plasmática del factor VIII, particularmente durante intervención quirúrgica.
Posología para hemorragia y cirugía: En caso de presentarse los siguientes eventos hemorrágicos, se debe mantener la actividad del factor VIII a los niveles plasmáticos o niveles superiores (en % del valor normal o en UI/dL) establecidos a continuación para el periodo de duración indicado.
|
Tipo de Hemorragia |
Nivel Requerido del Factor VIII (% o UI/dL) |
Frecuencia de las Dosis (h) |
|
Menor Hemartrosis temprana, hemorragias de músculos superficiales o tejido blando y hemorragias orales |
20-40 |
Repetir cada 12-24 horas de acuerdo con las necesidades hasta que se resuelva. Al menos 1 día, dependiendo de la severidad de la hemorragia. |
|
Moderada Hemorragia en los músculos. Traumatismo craneoencefálico leve. Operaciones menores que incluyen extracción de dientes. Hemorragias en la cavidad oral |
30-60 |
Infusión repetida cada 12-24 horas durante 3-4 días o hasta que la herida cure adecuadamente. |
|
Mayor Hemorragia gastrointestinal. Hemorragias intracraneales, intraabdominales o intratorácicas. Fracturas. Operaciones mayores. |
60-100 |
Infusión repetida cada 8-24 horas hasta que la amenaza se resuelva o en caso de cirugía hasta que sane adecuadamente la herida; se continúa con la terapia durante al menos otros 7 días. |
Posología para profilaxis: XYNTHA® se ha administrado profilácticamente en un estudio clínico principal en pacientes adolescentes y adultos con tratamiento previo a una dosis de 30 ± 5 UI/kg suministradas 3 veces a la semana.
Inhibidores: Los pacientes que utilizan terapia sustitutiva del factor VIII se deben controlar para determinar si se desarrollan inhibidores del factor VIII. Si los niveles plasmáticos esperados de la actividad del factor VIII no se obtienen o si la hemorragia no se controla con la dosis apropiada, se debe realizar un ensayo para determinar la presencia de inhibidores del factor VIII. En pacientes con inhibidores (especialmente con niveles altos de inhibidores, mayores de 5 Unidades Bethesda, UB), el tratamiento con el factor VIII podría no ser efectivo y deberán considerarse otras opciones terapéuticas. El manejo de estos pacientes debe ser dirigido por médicos con experiencia en el cuidado de pacientes con hemofilia. Ver también “PRECAUCIONES” y REACCIONES ADVERSAS”.
Administración: XYNTHA® Factor Antihemofílico (Recombinante), Libre de Plasma/Albúmina se administra por infusión intravenosa (IV) después de la reconstitución del polvo liofilizado con la jeringa prellenada suministrada con diluyente (solución de Cloruro de Sodio 0,9%, 4 mL). Si la solución y el contenedor lo permiten, los medicamentos parenterales se deben inspeccionar antes de la administración para determinar la presencia de material particulado y decoloración.
XYNTHA® Factor Antihemofílico (Recombinante), Libre de Plasma/Albúmina se debe administrar utilizando el juego de infusión suministrado en este kit y la jeringa prellenada de diluyente suministrada o una única jeringa plástica desechable estéril. Adicionalmente, la solución se debe extraer del vial utilizando el adaptador de vial.
1. Conecte la jeringa al extremo del equipo de infusión suministrado.
2. Coloque un torniquete y prepare el sitio de inyección limpiando bien la piel con una de las torundas de alcohol suministradas con el kit.
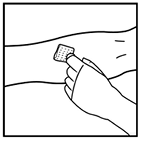
3. Realice la venopunción como se lo indicó su médico. Inserte la aguja del equipo de infusión dentro de la vena como se lo indicó su médico y retire el torniquete. Remueva el aire presente en el equipo de infusión retrayendo la jeringa. El producto XYNTHA® reconstituido se debe inyectar vía intravenosa durante varios minutos. La tasa de administración se debe determinar de acuerdo con el nivel de comodidad del paciente.
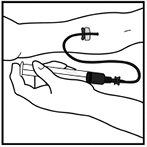
Luego de finalizar el tratamiento con XYNTHA®, retire el equipo de infusión y deséchelo. Descarte toda la solución no utilizada, los viales vacíos, las agujas y jeringas utilizadas depositándolos dentro de un recipiente apropiado para eliminación de desechos que pueden hacer daño a los demás si no se manipulan adecuadamente.
SOBREDOSIS: No se han reportado síntomas de sobredosis con productos del factor VIII de la coagulación recombinante.
INFORMACIÓN COMPLEMENTARIA
Descripción: El Factor Antihemofílico (Recombinante), Libre de Plasma/Albúmina, que es el ingrediente activo de XYNTHA®, es un factor VIII de la coagulación recombinante producido mediante tecnología de ADN recombinante para ser usado en el tratamiento de la deficiencia del factor VIII. El Factor Antihemofílico (Recombinante), Libre de Plasma/Albúmina es una glucoproteína purificada con una masa molecular aproximada de 170 kDa que se compone de 1.438 aminoácidos que no contienen el dominio B no activo. , La secuencia de aminoácidos de moroctocog alfa es similar a la forma 90 ? 80 kDa del factor VIII. Las modificaciones luego de las translación y las características funcionales in vitro de moroctocog alfa son similares a las del factor VIII endógeno.
El Factor Antihemofílico (Recombinante), Libre de Plasma/Albúmina es secretado por una línea celular del ovario del hámster chino (OHC) genomanipulada. La línea celular de OHC se ha estudiado extensamente y se ha determinado que no contiene virus detectables. La línea celular se somete a proliferación en un medio de cultivo químicamente definido que no contiene ningún material derivado de fuentes humanas o animales. El proceso de purificación se ha refinado para depurar la afinidad del moroctocog alfa empleando un método de cromatografía de columna que utiliza un ligando de afinidad sintetizado químicamente que reemplaza la resina Sepharose de anticuerpos monoclonales murinos y elimina la posibilidad de riesgo de contaminación viral asociada con el anticuerpo monoclonal murino y con su fabricación.
El Factor Antihemofílico (Recombinante), Libre de Plasma/Albúmina está completamente libre del riesgo de transmisión de patógenos por medio de la sangre humana, como por ejemplo el virus de la inmunodeficiencia humana (VIH), los virus de la hepatitis y el parvovirus, puesto que no se purifica a partir de sangre humana y se fabrica a partir de líneas celulares bien definidas sin presencia de materiales obtenidos de humanos o animales. Para mayor mejora del perfil de seguridad viral y para proporcionar seguridad adicional a la comunidad con hemofilia A, se han incluido durante la purificación un paso de desactivación viral con detergente solvente y un paso de nanofiltrado de retención de virus.
La proteína se purifica mediante un proceso de purificación que permite obtener un ingrediente activo de alta pureza. La potencia se expresa en unidades internacionales (UI), se determina utilizando un ensayo cromogénico de acuerdo con la Farmacopea Europea. El proceso de fabricación del estándar de trabajo para potencia de Wyeth se ha calibrado utilizando el ensayo de coagulación de una fase tomando como referencia la Séptima Norma Internacional de la Organización Mundial de la Salud (OMS) para actividad del factor VIII. Para ReFacto AF, la calibración del estándar para potencia se realiza utilizando el ensayo de sustrato cromogénico y para XYNTHA® se realiza la calibración del estándar para potencia. La actividad específica de XYNTHA® es de 5.500 - 9.900 UI por miligramo de proteína. La actividad específica de ReFacto AF es 7600 - 13800 UI por miligramo de proteína.
Es posible que la información de prescripción de este producto haya sido revisada y actualizada después de la fecha de impresión del PLM 2016. Para obtener información más actualizada comuníquese con la Dirección Médica de Pfizer S.A.S Teléfono: (1) 6002300 Ext. 2509 Bogotá – Colombia.
DESCRIPCIÓN: XYNTHA® es un polvo liofilizado blanco a blancuzco. Después de reconstituido, la apariencia de XYNTHA® es la de una solución incolora clara a levemente opalescente.
PRESENTACIÓN: XYNTHA® 250 I.U. Polvo liofilizado para solución inyectable (Reg. San. INVIMA 2009M-0009692). XYNTHA® 500 I.U. Polvo Liofilizado para Solución Inyectable (Reg. San. INVIMA 2009M-0009675).
XYNTHA® 1000 I.U. Polvo liofilizado para solución inyectable (Reg. San. INVIMA2012M-0013847). XYNTHA® 2000 I.U. (Reg. San. INVIMA 2012M-0013753)
Título del Documento de Producto: Factor Antihemofílico (Recombinante), libre de Plasma/Albúmina.
Fecha de CDS que reemplaza: Agosto 23, 2011
Fecha Efectiva: Enero 09, 2012
Versión CDS: 12.0
ALMACENAMIENTO Y VIDA ÚTIL: Producto en el empaque de venta: XYNTHA® se debe almacenar bajo refrigeración a una temperatura de 2? a 8?C (36? a 46?F) por hasta 3 años. Se deberá evitar congelarlo para evitar el daño de la jeringa prellenada de diluyente. Durante el almacenamiento, se debe evitar la exposición prolongada a la luz del vial de XYNTHA®.
Producto después de reconstituido: El producto no contiene preservantes y deberá utilizarse inmediatamente después de reconstituido.
ELIMINACIÓN: Todos los productos no utilizados o el material de desperdicio se deben eliminar de acuerdo con los requisitos locales.
MANIPULACIÓN: XYNTHA®, cuando se reconstituye, contiene polisorbato-80, que se conoce aumenta la tasa de extracción de di-(2-etilexil) ftalato (DEHP) del cloruro de polivinilo (PVC). Esto debe ser considerado durante la preparación y la administración de XYNTHA®, incluyendo el tiempo de almacenamiento transcurrido en el envase de PVC después de la reconstitución. Es importante que las recomendaciones de posología y administración sean seguidas de manera estricta.
Reconstitución: Lávese siempre las manos antes de realizar los siguientes procedimientos. Durante el procedimiento de reconstitución se debe utilizar técnica aséptica (es decir bajo condiciones de limpieza y sin gérmenes). Para minimizar su exposición innecesaria a la atmósfera, todos los componentes utilizados en la reconstitución y administración de este producto se deben utilizar tan pronto como sea posible después de abrir sus contenedores estériles.
XYNTHA® Factor Antihemofílico (Recombinante), Libre de Plasma/Albúmina se administra vía infusión intravenosa (IV) después de la reconstitución con la jeringa de diluyente suministrada (solución de cloruro de sodio 0,9%).
Nota: Si utiliza más de un vial de XYNTHA® por infusión, cada vial se debe reconstituir de acuerdo con las siguientes instrucciones. La jeringa de diluyente se debe retirar, dejando el adaptador vial en su lugar y se puede utilizar una única jeringa grande para extraer el contenido reconstituido de cada uno de los viales individuales. No suelte las jeringas de diluyente o la jeringa grande hasta que esté listo para colocar la jeringa grande al siguiente adaptador para vial.
1. Deje que los viales de XYNTHA® liofilizados y la jeringa prellenada de diluyente alcance la temperatura ambiente.
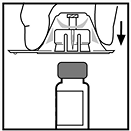
2. Retire la tapa plástica de cierre del vial de XYNTHA® para que quede visible la parte central del tapón de caucho.
3. Limpie la parte superior del vial con la torunda de alcohol suministrada o utilice otra solución antiséptica, deje que se seque. Después de limpiar el tapón de caucho, no lo toque con su mano ni permita que toque ninguna superficie.
4. Desprenda la tapa del empaque del adaptador de vial plástico transparente. No retire el adaptador del empaque.
5. Coloque el vial en una superficie plana. Mientras sujeta el empaque del adaptador, coloque el adaptador de vial sobre el vial y presione firmemente hacia abajo sobre el empaque hasta que el punzón penetre en el tapón del vial. Deje en su lugar el empaque del adaptador.
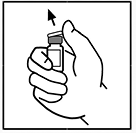
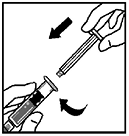
6. Tome la barra del émbolo como se presenta en la figura. Evite tocar el eje de la barra del émbolo. Ensamble el extremo roscado de la barra del émbolo a la jeringa de diluyente insertando la barra dentro de la abertura del tapón de la jeringa y empujando y girando firmemente la barra hasta que se asegure al tapón.
7. Rompa la tapa de la punta de cierre inviolable de la jeringa de diluyente partiendo la perforación de la tapa. Esto se puede realizar doblando la tapa hacia arriba y hacia abajo hasta que la perforación se rompa. No toque el interior de la tapa o la punta de la jeringa. Podría ser necesario volver a colocar la tapa (sino se administra inmediatamente XYNTHA®, por ello se recomienda poner la tapa a un lado colocándola apoyada sobre su parte superior.
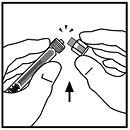
8. Retire el empaque del adaptador y deséchelo.
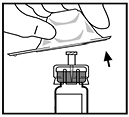
9. Coloque el vial sobre una superficie plana. Conecte la jeringa del diluyente al adaptador del vial insertando la punta de la jeringa dentro de la abertura del adaptador y al mismo tiempo empuje firmemente y gire la jeringa en dirección de las manecillas del reloj hasta que asegure la unión.
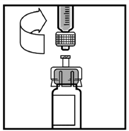
10. Empuje lentamente la barra del émbolo para inyectar todo el diluyente dentro del vial de XYNTHA®.
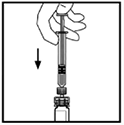
11. Sin retirar la jeringa, rote suavemente el vial hasta que el polvo se disuelva.
Nota: Antes de la administración la solución final se debe inspeccionar visualmente para determinar si existe material particulado. La solución debe lucir clara a levemente opalescente e incolora. Si no es así, se debe descartar la solución y utilizar un nuevo kit.
12. Asegúrese que la barra del émbolo de la jeringa se encuentra todavía completamente oprimida, invierta el vial y suavemente extraiga dentro de la jeringa toda la solución a través del adaptador para vial.
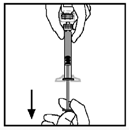
13. Suelte la jeringa del adaptador del vial jalando y girando suavemente la jeringa en dirección contraria de las manecillas del reloj. Deseche el vial con el adaptador puesto.
Nota: Si no se va a utilizar inmediatamente la solución, la tapa de la jeringa se debe volver a colocar cuidadosamente. No toque la punta de la jeringa o el interior de la tapa.
REFERENCIAS
3.2.P.5.1, Table 1-1
3.2.S.1.1, Table 1-1
WHO Collaborating Centre for Drug Statistics Methodology. Available at: http://www.whocc.no/atcddd/. Last accessed 25 May 2007.
3.2.P.1, Section 1.1
3.2.P.1, Section 2.0, Table 2-1
3.2.P.1, Section 2.0, Table 2-1
2.7.3, Section 3.2
3.2.S.1.3, Section 3.0
2.7.3, Section 3.2.2.1.3
2.7.3, Section 3.2.2.1.4
2.7.3, Section 3.2.2.1.3
2.7.3, Section 3.2.2.1.4
Nilsson IM, Berntorp EE and Freiburghaus C. Treatment of patients with factor VIII and IX inhibitors. Thromb Haemost. 1993;70(1):56-59.
Hoyer LW. Hemophilia A. N Engl J Med. 1994;330:38-47.
EMEA Committee for Proprietary Medicinal Products: Core SPC for Human Plasma Derived and Recombinant Coagulation Factor VIII Products. London, 29 June 2000. Doc. Ref. CPMP/BPWG/1619/99.
3.2.P.2.3, Section 3.3.1
3.2.P.6
EMEA Committee for Proprietary Medicinal Products: Core SPC for Human Plasma Derived and Recombinant Coagulation Factor VIII Products. London, 29 June 2000. Doc. Ref. CPMP/BPWG/1619/99.
2.7.3, Section 3.2.1.1.1
White GC II, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J, on behalf of the Factor VIII and Factor IX Subcommittee. Scientific and Standardization Committee Communication: Definitions in Hemophilia: Recommendation of the Scientific Subcommittee on Factor VIII and Factor IX of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 2001;85:560.
Nilsson IM, Berntorp EE and Freiburghaus C. Treatment of patients with factor VIII and IX inhibitors. Thromb Haemost. 1993;70(1):56-59.
3.2.P.1, Section 1.1
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in hemophiliacs. Lancet. 1992;339:594 598.
Lusher J, Arkin S, Abildgaard CF, Schwartz RS, Group TKPUPS. Recombinant factor VIII for the treatment of previously untreated patients with hemophilia A. N Engl J Med. 1993;328:453-459.
Bray GL, Gomperts ED, Courter S, et al. A multicenter study of recombinant factor VIII (Recombinate): safety, efficacy, and inhibitor risk in previously untreated patients with hemophilia A. Blood. 1994;83(9):2428-2435.
Kessler C, Sachse K. Factor VIII:C inhibitor associated with monoclonal-antibody purified FVIII concentrate. Lancet. 1990;335:1403.
Schwartz RS, Abildgaard CF, Aledort LM, et al. Human recombinant DNA-derived antihemophilic factor (factor VIII) in the treatment of hemophilia A. N Engl J Med. 1990;323:1800-1805.
White GC II, Courter S, Bray GL, et al. A multicenter study of recombinant factor VIII (recombinate) in previously treated patients with hemophilia A. Thromb Haemost. 1997;77(4):660-667.
Gruppo R, Chen H, Schroth P, et al. Safety and immunogenicity of recombinant factor VIII (Recombinate) in previously untreated patients: A 7.3 year update. Haemophilia. 1998;4:228 (Abstract No. 291, XXIII Congress of the WFH, The Hague).
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in haemophilia A patients - a review of recent studies of recombinant and plasma-derived factor VIII concentrates. Haemophilia. 1999;5:145-154.
Abshire TC, Brackmann HH, Scharrer I, et al. Sucrose formulated recombinant human antihemophilic Factor VIII is safe and efficacious for treatment of hemophilia A in home therapy: Results of a multicenter, international, clinical investigation. Thromb Haemost. 2000;83(6):811-816.
2.7.4, Section 5.1
5.3.3.2.1, CSR-66997, Section 11.1
3.2.S.2.6, Section 3.0
5.3.5.4, CSR-49151, Section 8.1
Courter SG, Bedrosian CL: Clinical evaluation of B-domain deleted recombinant factor VIII in previously untreated patients. Semin Hematol. 2001;38(2 Suppl 4):52-59.
Courter SG, Bedrosian CL: Clinical evaluation of B-domain deleted recombinant factor VIII in previously treated patients. Semin Hematol. 2001;38(2 Suppl 4):44-51.
Lusher JM, Lee CA, Kessler CM and Bedrosian CL: The safety and efficacy of B-domain deleted recombinant factor VIII concentrate in Patients with severe Haemophilia A. Haemophilia. 2003; 9: 38 49.
Lusher JM, Roth DA. The safety and efficacy of B-domain deleted recombinant factor VIII concentrates in patients with severe Haemophilia A: an update. Haemophilia. 2005;11:292-293.
2.7.4, Section 1.3
EMEA. Committee for Proprietary Medicinal Products (CMPC). Core SPC for Humane Plasma Derived and Recombinant Coagulation Factor VIII Products. Canary Wharf, London. 2007;p7
EMEA. Committee for Proprietary Medicinal Products (CMPC). Core SPC for Humane Plasma Derived and Recombinant Coagulation Factor VIII Products. Canary Wharf, London. 2007;p7
(310)CSR-66997: Final report: a randomized two-way blinded crossover-design study to establish the bioequivalence of B-domain deleted recombinant factor VIII (BDDrFVIII, moroctocog alfa [AF-CC]) with a full-length recombinant factor VIII preparation (FLrFVIII, Advate), followed by an open-label trial of the safety and efficacy of moroctocog alfa [AF-CC] in previously treated patients with hemophilia A. Version 1.0, dated 13-Mar-2007.
(306)CSR-58042: Final report: an open-label study to characterize the safety and efficacy of B-domain deleted recombinant factor VIII (BDDrFVIII) and manufactured by the albumin-free process (ReFacto AF) in the treatment of previously treated patients with severe hemophilia A. Version 1.0, dated 27-Mar-2006.
Study 307
Recht M, Nemes L, Matsiak M, et al. Clinical evaluation of moroctocog alfa (AF-CC), a new generation of B-domain deleted recombinant factor VIII (BDDrFVIII) for treatment of haemophilia A: demonstration of safety, efficacy, and pharmacokinetic equivalence to full length recombinant factor VIII. Haemophilia. 2009;15:869-880.
Study 300
(310)CSR-66997: Final report: a randomized two-way blinded crossover-design study to establish the bioequivalence of B-domain deleted recombinant factor VIII (BDDrFVIII, moroctocog alfa [AF-CC]) with a full-length recombinant factor VIII preparation (FLrFVIII, Advate), followed by an open-label trial of the safety and efficacy of moroctocog alfa [AF-CC] in previously treated patients with hemophilia A. Version 1.0, dated 13-Mar-2007.
2.7.4, Section 2.1
Recht M, Nemes L, Matsiak M, et al. Clinical evaluation of moroctocog alfa (AF-CC), a new generation of B-domain deleted recombinant factor VIII (BDDrFVIII) for treatment of haemophilia A: demonstration of safety, efficacy, and pharmacokinetic equivalence to full length recombinant factor VIII. Haemophilia. 2009;15:869-880.
Study 311 CSR from Dec 2009.
CSR-66997: Final report: a randomized two-way blinded crossover-design study to establish the bioequivalence of B-domain deleted recombinant factor VIII (BDDrFVIII, moroctocog alfa [AF-CC]) with a full-length recombinant factor VIII preparation (FLrFVIII, Advate), followed by an open-label trial of the safety and efficacy of moroctocog alfa [AF-CC] in previously treated patients with hemophilia A. Version 1.0, dated 13-Mar-2007.
CSR-58042: Final report: an open-label study to characterize the safety and efficacy of B-domain deleted recombinant factor VIII (BDDrFVIII) and manufactured by the albumin-free process (ReFacto AF) in the treatment of previously treated patients with severe hemophilia A. Version 1.0, dated 27 Mar 2006.
Mann KG and Ziedens KB. Overview of Hemostasis. In: Lee CA, Berntorp EE and Hoots WK , eds. Textbook of Hemophilia. USA, Blackwell Publishing; 2005: 1-4.
Mann KG and Ziedens KB. Overview of Hemostasis. In: Lee CA, Berntorp EE and Hoots WK, eds. Textbook of Hemophilia. USA, Blackwell Publishing; 2005: 1-4.
Mann KG and Ziedens KB. Overview of Hemostasis. In: Lee CA, Berntorp EE and Hoots WK, eds. Textbook of Hemophilia. USA, Blackwell Publishing; 2005: 1-4.
2.7.2, Section 2.1.2
3.2.S.1.2
Kaufman, R. Cellular processing of factors VIII and IX. In: Lee CA, Berntorp EE and Hoots WK , eds. Textbook of Hemophilia. USA, Blackwell Publishing; 2005:5-12.
3.2.P.1, Section 1.1
Mann KG and Ziedens KB. Overview of Hemostasis. In: Lee CA, Berntorp EE and Hoots WK, eds. Textbook of Hemophilia. USA, Blackwell Publishing; 2005: 1-4.
Mann KG and Ziedens KB. Overview of Hemostasis. In: Lee CA, Berntorp EE and Hoots WK, eds. Textbook of Hemophilia. USA, Blackwell Publishing; 2005: 1-4.
Mann KG and Ziedens KB. Overview of Hemostasis. In: Lee CA, Berntorp EE and Hoots WK, eds. Textbook of Hemophilia. USA, Blackwell Publishing; 2005: 1-4.
Mann KG and Ziedens KB. Overview of Hemostasis. In: Lee CA, Berntorp EE and Hoots WK, eds. Textbook of Hemophilia. USA, Blackwell Publishing; 2005: 1-4.
5.3.3.2.1, CSR 66997, Section 6.1
2.7.3, Section 2.1
2.7.1, Section 2.2.1.3
2.7.2, Section 2.1.2
2.7.3, Section 2.1
2.7.3, Section 3.2.1.1.1
2.7.3, Section 3.2.1.1.2
2.7.3, Section 3.2.1.1.2
2.7.3 section 3.2.2.1.1
2.7.3, section 3.2.2.1.3
2.7.3, Section 3.2.1.1.4
2.7.3, Section 3.2.2.1.5.1
2.7.4, Section 1.2
2.7.3, Section 2.3
2.7.3, Section 2.3
2.7.3, Section 2.3
5.3.3.2.1, CSR-66997, Section 9.3.1
5.3.3.2.1, CSR-66997, Section 9.3.1
5.3.3.2.1, CSR-66997, Section 9.3.2
2.7.2, Section 2.1.2
5.3.3.2.1, CSR-66997, Section 9.4.2
2.7.2, Section 2.1.2
2.7.2, Section 2.1.2
2.7.2, Section 2.1.2
2.7.2, Section 2.1.2
5.3.3.2.1, CSR-66997, Section 9.4.2
5.3.5.2, Section 1.0, Pharmacokinetics, Table 1-6, Study 3082B2-311-WW – Progress Report
PUP CSR: Skillman CA, Huang D, Maher T. Clinical Safety and Efficacy Study of Recombinant Factor VIII (BDDrFVIII) During Long-Term Prophylaxis and/or On-Demand Treatment in Previously Untreated Patients (PUPs) With Severe Hemophilia A: Final Report. Protocols 3082A1-301-WW.
PTP CSR: Davis M, Huang D, Maher T. Clinical Safety and Efficacy Studies of Recombinant Factor VIII (BDDrFVIII, formerly r-VIII SQ), Pharmacia & Upjohn AB Sweden, During Long-Term Prophylaxis and/or On-Demand Treatment in Previously Treated Hemophilia A Patients: Final Report. Protocols 3082A1-300-WW.
PUP CSR: Skillman CA, Huang D, Maher T. Clinical Safety and Efficacy Study of Recombinant Factor VIII (BDDrFVIII) During Long-Term Prophylaxis and/or On-Demand Treatment in Previously Untreated Patients (PUPs) With Severe Hemophilia A: Final Report. Protocols 3082A1-301-WW.
PUP CSR: Skillman CA, Huang D, Maher T. Clinical Safety and Efficacy Study of Recombinant Factor VIII (BDDrFVIII) During Long-Term Prophylaxis and/or On-Demand Treatment in Previously Untreated Patients (PUPs) With Severe Hemophilia A: Final Report. Protocols 3082A1-301-WW.
3.2.S.2.6
2.4
2.6.6, Section 4.0
2.4 Section 3.1
4.2.1.1 RPT 43756
2.4, Section 5.0
4.2.3.2, RPT-41791
4.2.3.2, RPT-66704
Juhlin F. Stability and Compatibility of Reconstituted Recombinant Factor VIII SQ, 250 IU/ml, in a System for Continuous Infusión. Pharmacia Document 9610224, 1996.
3.2.P.8.1
3.2.P.8.1, Section 2.0
3.2.P.8.1
3.2.P.1, Section 1.1
3.2.S.1.2
Kaufman, R. Cellular processing of factors VIII and IX. In: Lee CA, Berntorp EE and Hoots WK , eds. Textbook of Hemophilia. USA, Blackwell Publishing; 2005:5-12.
3.2.S.1.2
3.2.S.1.2
3.2.S.1.3
3.2.S.1.3
3.2.S.2.3, Section 4.0
3.2.S.2.3
3.2.S.2.2, Section 5.4
3.2.S.2.2, Section 5
3.2.S.4.4
3.2.P.5.2.1
3.2.P.2.3, Section 3.3.1
3.2.P.5.1
PFIZER S.A.S.


