

ZYTIGA
ABIRATERONA
Tabletas
Caja, 1 Frasco(s), 120 Tabletas,
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA: Las TABLETAS de ZYTIGA® contienen 250 mg de acetato de abiraterona.
INDICACIONES:
ZYTIGA® en combinación con prednisona o prednisolona, está indicado para: El tratamiento de pacientes con cáncer de próstata metastásico resistente a la castración (mCRPC, por sus siglas en inglés) que sean asintomáticos o levemente sintomáticos, tras el fracaso con el tratamiento de privación de andrógenos.
El tratamiento del cáncer de próstata metastásico avanzado (cáncer de próstata resistente a la castración) en pacientes que han recibido quimioterapia previa conteniendo un taxano.
ZYTIGA® está indicado en combinación con prednisona o prednisolona y con terapia de privación de andrógenos (ADT, por sus siglas en inglés) para el tratamiento de cáncer de próstata metastásico de alto riesgo sin tratamiento hormonal previo (mHNCP, por sus siglas en inglés) o para el tratamiento de cáncer de próstata metastásico sensible a hormonas (mHSPC, por sus siglas en inglés) de diagnosticado reciente de alto riesgo.
FORMA FARMACÉUTICA: Tabletas. Venta bajo receta médica.
INFORMACIÓN FARMACÉUTICA:
Lista de excipientes: ZYTIGA® 250 mg tableta no recubierta contiene los siguientes excipientes:
Dióxido de silicio coloidal, croscarmelosa sódica, lactosa monohidrato, estearato de magnesio, celulosa microcristalina, povidona, lauril sulfato de sodio.
Incompatibilidades: No aplicable.
Período de validez: Observar la fecha de caducidad en el envase exterior.
PROPIEDADES FARMACOCINÉTICAS:
Introducción general: Después de la administración de acetato de abiraterona, se ha estudiado la farmacocinética de abiraterona y acetato de abiraterona en sujetos sanos, en pacientes con cáncer de próstata metastásico avanzado y en sujetos sin cáncer con insuficiencia hepática o renal. El acetato de abiraterona se convierte rápidamente in vivo a abiraterona, un inhibidor de la biosíntesis de andrógenos (ver sección Propiedades Farmacológicas - Mecanismo de acción).
Absorción: Después de la administración de acetato de abiraterona por vía oral en ayuno, el tiempo hasta alcanzar la máxima concentración plasmática de abiraterona es de aproximadamente 2 horas.
La administración de acetato de abiraterona con los alimentos, en comparación con la administración en ayunas, da como resultado un incremento de hasta 17 veces la exposición sistémica media de abiraterona dependiendo del contenido graso de la comida. Dada la variación normal del contenido y la composición de las comidas, la ingesta de ZYTIGA® con los alimentos tiene el potencial de producir exposiciones muy variables. Por tanto, ZYTIGA® no se debe tomar con los alimentos. ZYTIGA® se debe tomar al menos dos horas después de comer y no se debe ingerir alimentos durante al menos una hora después de tomar ZYTIGA®. Las tabletas se deben ingerir enteras con agua (ver sección Posología y administración).
Distribución y unión a las proteínas: La unión a las proteínas plasmáticas de la 14C-abiraterona en el plasma humano es 99.8%. El volumen aparente de distribución es aproximadamente 5630 L, sugiendo que la abiraterona se distribuye ampliamente en los tejidos periféricos.
Metabolismo: Después de la administración oral de acetato de 14C-abiraterona en cápsulas, el acetato de abiraterona se hidroliza a abiraterona, que posteriormente se metaboliza, que luego experimenta metabolismo incluyendo sulfatación, hidroxilación y oxidación, principalmente en el hígado. La mayor parte de la radiactividad circulante (aproximadamente 92%) se encuentra en forma de metabolitos de abiraterona. De 15 metabolitos detectables, 2 metabolitos principales,sulfato de abiraterona y N-óxido de sulfato de abiraterona, representan cada uno aproximadamente el 43% de la radiactividad total.
Eliminación: La semivida media de abiraterona en el plasma es aproximadamente 15 horas basado en los datos de los sujetos sanos. Después de la administración de acetato de 14C-abiraterona, aproximadamente el 88% de la dosis radiactiva se recupera en las heces y aproximadamente el 5% en la orina. Los compuestos principales presentes en las heces son acetato de abiraterona inalterado y abiraterona (aproximadamente 55% y 22% de la dosis administrada, respectivamente).
Poblaciones especiales:
Insuficiencia renal: Se comparó la farmacocinética de abiraterona en pacientes con enfermedad renal en etapa terminal sometidos a un programa estable de hemodiálisis frente a sujetos control emparejados con función renal normal. La exposición sistémica a abiraterona después de una única dosis oral de 1000 mg no se incrementó en los pacientes con enfermedad renal en etapa terminal sometidos a diálisis.
La administración de ZYTIGA® en pacientes con insuficiencia renal incluyendo la insuficiencia renal severa, no requiere reducción de la dosis (ver sección Posología y administración - Insuficiencia renal).
Insuficiencia hepática: Se examinó la farmacocinética de abiraterona en sujetos con insuficiencia hepática leve o moderada preexistente (Clase A y B de Child-Pugh, respectivamente) y en sujetos sanos de control. La exposición sistémica a abiraterona después de una única dosis oral de 1000 mg se incrementó en aproximadamente 11% y 260% en sujetos con insuficiencia hepática leve y moderada preexistente, respectivamente. La semivida media de abiraterona se prolonga hasta aproximadamente 18 horas en sujetos con insuficiencia hepática leve y hasta aproximadamente 19 horas en sujetos con insuficiencia hepática moderada. No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve preexistente. No existen datos sobre seguridad y eficacia clínica de las dosis múltiples de acetato de abiraterona cuando se administró a pacientes con insuficiencia hepática moderada o severa (Clase B o C de Child-Pugh). No se puede predecir el ajuste de la dosis. ZYTIGA® se debe usar con precaución en pacientes con insuficiencia hepática moderada, solo si el beneficio claramente supera el posible riesgo (ver sección Posología y administración -Insuficiencia hepática y Advertencias y precauciones - Hepatotoxicidad e Insuficiencia hepática). ZYTIGA® no se debe utilizar en pacientes con insuficiencia hepática severa. Puede ser necesario ajustar la dosis o suspender el tratamiento en pacientes que desarrollan hepatotoxicidad durante el tratamiento con ZYTIGA® (ver sección Posología y administración - Insuficiencia hepática y Advertencias y precauciones - Hepatotoxicidad e Insuficiencia hepática).
Efectos en el intervalo QT:
En un estudio de seguridad cardiovascular en pacientes con cáncer de próstata metastásico avanzado no hubo efectos significantes de acetato de abiraterona en el intervalo QT/QTc cardiaco.
PROPIEDADES FARMACODINÁMICAS:
Grupo farmacoterapéutico: Otros antagonistas hormonales y agentes relacionados, código ATC: L02BX03.
Mecanismo de acción: El acetato de abiraterona (ZYTIGA®) se convierte in vivo a abiraterona, un inhibidor de la biosíntesis de andrógenos. Especificamente, la abiraterona inhibe selectivamente la enzima 17a-hidroxilasa/C17,20-liasa (CYP17). Esta enzima se expresa y es necesaria para la biosíntesis de andrógenos en los tejidos tumorales testiculares, suprarrenales y prostáticos. Cataliza la conversión de pregnenolona y progesterona a los precursores de la testosterona, DHEA y androstenediona, respectivamente, mediante 17a-hidroxilación y rotura del enlace C17,20. La inhibición de CYP17 también produce incremento de mineralocorticoides por las glándulas suprarrenales (ver sección Advertencias y precauciones - Hipertensión, hipocalemia y retención de líquidos debido al exceso de mineralocorticoides).
El carcinoma de próstata sensible a los andrógenos responde al tratamiento que disminuye los niveles de andrógenos. Las terapias de privación de andrógenos, como el tratamiento con agonistas de LHRH u orquiectomía, disminuyen la producción testicular de andrógenos pero no afectan la producción de andrógenos por las glándulas suprarenales o en el tumor El tratamiento con ZYTIGA® disminuye la testosterona sérica hasta niveles indetectables (utilizando ensayos comerciales) cuando se administra con agonistas de LHRH (u orquiectomía).
CONTRAINDICACIONES: Hipersensibilidad al acetato de abiraterona o alguno de los excipientes.
Embarazo: ZYTIGA® está contraindicado en mujeres que están o podrían potencialmente embarazarse (ver sección Embarazo, lactancia y fertilidad - Embarazo).
EMBARAZO, LACTANCIA Y FERTILIDAD:
Embarazo: ZYTIGA® está contraindicado en mujeres que están embarazadas o pueden potencialmente embarazarse (ver sección Contraindicaciones).
No existen datos en humanos sobre el uso de ZYTIGA® en el embarazo y ZYTIGA® no es para uso en mujeres en edad fértil. Se espera que el uso materno de un inhibidor del CYP17 produzca cambios en los niveles hormonales que pueden afectar el desarrollo del feto (ver sección Propiedades farmacológicas - Mecanismo de acción e Información No-clínica - Toxicología reproductiva).
Se desconoce si la abiraterona o sus metabolitos están presentes en el semen. Se requiere el uso de un preservativo si el paciente tiene actividad sexual con una mujer embarazada. Si el paciente tiene actividad sexual con una mujer en edad fértil, se requiere el uso de un preservativo junto con otro método anticonceptivo efectivo.
Para evitar la exposición involuntaria, las mujeres embarazadas o las que puedan estar embarazadas no deben manipular las tabletas no recubiertas de ZYTIGA® 250 mg sin protección, por ejemplo, guantes.
Lactancia: ZYTIGA® no es para uso en mujeres.
No se sabe si el acetato de abiraterona o sus metabolitos se segregan en la leche materna humana.
Efectos en la capacidad para conducir y utilizar máquinas: No se han realizado estudios sobre los efectos de ZYTIGA® en la capacidad para conducir o utilizar máquinas. No se espera que ZYTIGA® afecte la capacidad para conducir y utilizar máquinas.
EFECTOS FARMACODINÁMICOS: ZYTIGA® disminuye la testosterona sérica y otros andrógenos hasta niveles inferiores a los alcanzados con el uso solo de agonistas de LHRH o con orquiectomía. Esto es resultado de la inhibición selectiva de la enzima CYP17 necesaria para la biosíntesis de androgénos. El antígeno prostático específico (PSA, por sus siglas en Inglés) sirve como biomarcador en pacientes con cáncer de próstata. En un estudio clínico de fase 3 de pacientes que fracasaron con la quimioterapia previa con taxanos, el 38% de los pacientes tratados con ZYTIGA® frente al 10% de los pacientes tratados con el placebo, presentaron al menos una reducción del 50% de los niveles del PSA desde el valor basal.
Uso de espironolactona:A los pacientes en los estudios clínicos pivotales con ZYTIGA® no se les permitió usar espironolactona ya que la espironolactona se une al receptor de andrógenos y puede incrementar los niveles del PSA.
REACCIONES ADVERSAS: En esta sección se presentan las reacciones adversas. Las reacciones adversas son eventos adversos que se consideraron estar razonablemente asociados con el uso de acetato de abiraterona basado en la evaluación exhaustiva de la información disponible del evento adverso. Una relación causal con acetato de abiraterona no puede ser establecida de manera confiable en casos individuales. Además, debido a que los estudios clínicos son conducidos bajo condiciones muy variantes, las tasas observadas de la reacción adversa en los estudios clínicos de un fármaco no pueden ser directamente comparadas con las tasas en los estudios clínicos de otro fármaco y puede no reflejar las tasas observadas en la práctica clínica.
En un análisis de la reacción adversa de los estudios compuesto de Fase 3 con ZYTIGA®, las reacciones adversas que se observaron en ≥ 10% de los pacientes fueron hipertensión, edema periférico, hipocalemia, infección del tracto urinario e incremento de la aspartato aminotransferasa y/o incremento de la alanina aminotransferasa.
ZYTIGA® puede causar hipertensión, hipocalemia y retención de líquidos como una consecuencia farmacodinámica de su mecanismo de acción. En estudios de fase 3, los efectos mineralocorticoides esperados se observaron con más frecuencia en pacientes tratados con ZYTIGA® frente a los pacientes tratados con el placebo: la hipocalemia 18% frente al 8%, hipertensión 22% frente al 16% y retención de líquidos (edema periférico) 23% frente al 17%, respectivamente. En los pacientes tratados con ZYTIGA®, se observó hipocalemia de grado 3 y 4 en el 6% y el 2% de los pacientes, hipertensión de grado 3 y 4 en el 8% y el 5% de los pacientes y edema por retención de líquidos de grado 3 y 4 en el 1% y el 1% de los pacientes, respectivamente. Los efectos mineralocorticoides generalmente pudieron ser manejados con éxito en términos médicos. El uso concomitante de un corticosteroide reduce la incidencia y la severidad de estas reacciones adversas (ver sección Advertencias y precauciones - Hipertensión, hipocalemia y retención de líquidos debido a exceso de mineralocorticoides).
En estudios de pacientes con cáncer de próstata metastásico avanzado que estaban utilizando un agonista de la hormona liberadora de hormona luteinizante (LHRH) o que fueron tratados previamente con orquiectomía, se administró ZYTIGA® a una dosis diaria de 1000 mg en combinación con prednisona o prednisolona a dosis bajas (5 o 10 mg diarios).
En la tabla 1 se muestran las reacciones adversas que ocurrieron con una frecuencia de ≥ 1% (todos los grados):
|
Tabla 1: Reacciones adversas en ≥ 1% de los pacientes en los estudios clínicosa |
|||
|
|
1000 mg de ZYTIGA® diariamente con prednisona o prednisolona n = 2659b |
||
|
Sistema de clasificación de órganos Reacción adversa |
Todos los grados % |
Grado 3 % |
Grado 4 % |
|
Trastornos generales y condiciones en el lugar de administración |
|||
|
Edema periférico |
20 |
< 1 |
0 |
|
Trastornos del metabolismo y de la nutrición |
|||
|
Hipocalemia |
20 |
5 |
<1 |
|
Hipertrigliceridemia |
1 |
0 |
0 |
|
Infecciones e infestaciones |
|||
|
Infección del tracto urinario |
10 |
2 |
<1 |
|
Trastornos hepatobiliares |
|||
|
ALT incrementada y/o AST incrementadac |
13 |
4 |
< 1 |
|
Trastornos vasculares |
|||
|
Hipertensión |
21 |
6 |
0 |
|
Lesiones, intoxicaciones y complicaciones de procedimiento |
|||
|
Fracturasd |
7 |
2 |
<1 |
|
Trastornos cardiacos |
|||
|
Insuficiencia cardiacae |
1 |
< 1 |
<1 |
|
Angina de pecho |
2 |
<1 |
0 |
|
Arritmia |
1 |
0 |
0 |
|
Fibrilación auricular |
3 |
1 |
<1 |
|
Taquicardia |
2 |
<1 |
0 |
|
Trastornos renales y urinarios |
|||
|
Hematuria |
7 |
1 |
0 |
|
Trastornos gastrointestinales |
|||
|
Dispepsia |
6 |
0 |
0 |
|
a Todos los pacientes estaban utilizando un agonista de la LHRH o se habían sometido a una orquiectomía. b n = pacientes en los que se ha evaluado la seguridad c ALT incrementado y/o AST incrementado incluye ALT incrementado, AST incrementado y función anormal hepática. d Fracturas incluye osteoporosis y todas las fracturas, a excepción de las fracturas patológicas e Insuficiencia cardiaca incluye insuficiencia cardiaca congestiva, disfunción venticular izquierda y disminución de la fracción de eyección |
|||
La reacción adversa, insuficiencia suprarrenal, ocurrió en los estudios clínicos de fase 3 a una tasa de 0.3% en los pacientes que recibieron ZYTIGA® y con una tasa del 0.1% en los pacientes que recibieron el placebo.
Efectos cardiovasculares: Los tres estudios de Fase 3 excluyeron a los pacientes con hipertensión no controlada, enfermedad cardiaca clínicamente significativa como el evidenciado por infarto de miocardio, eventos trombóticos arteriales en los últimos 6 meses, angina severa o inestable, insuficiencia cardiaca de clase III o IV de la NYHA (Estudio 301) o insuficiencia cardíaca de clase II a IV (Estudios 3011 y 302) o medición de la fracción de eyección cardiaca < 50%. Todos los pacientes enrolados (pacientes tratados con el activo y con el placebo) fueron tratados concomitantemente con terapia de privación de andrógenos, predominantemente con el uso de agonistas de la LHRH, la cual se ha asociado con diabetes, infarto de miocardio, accidente cerebrovascular y muerte cardiaca súbita. La incidencia de reacciones adversas cardiovasculares en los estudios de fase 3 en los pacientes que recibieron ZYTIGA® frente a los que recibieron el placebo fue la siguiente: fibrilación auricular 2.6% frente al 2.0%, taquicardia 1.9% frente al 1.0%, angina de pecho: 1.7% frente al 0.8%, insuficiencia cardíaca 0.7% frente al 0.2% y arri™ia 0.7% frente al 0.5%.
Hepatotoxicidad: Se han reportado hepatotoxicidad asociada al fármaco con la elevación de ALT, AST y bilirrubina total en pacientes tratados con ZYTIGA®. A lo largo de los estudios clínicos de fase 3 se ha reportado hepatotoxicidad de grado 3 y 4 (por ejemplo, incrementos de ALT o AST > 5 X LSN o incrementos de bilirrubina > 1.5 X LSN) en aproximadamente 6% de los pacientes que recibieron ZYTIGA®, típicamente durante los 3 primeros meses después de iniciar el tratamiento. En el Estudio 3011, se observó hepatotoxicidad de grado 3 o 4 en el 8.4% de los pacientes tratados con ZYTIGA®. Diez pacientes que recibieron ZYTIGA® fueron discontinuados debido a hepatotoxicidad; dos presentaron hepatotoxicidad de grado 2, seis presentaron hepatotoxicidad de grado 3 y dos presentaron hepatotoxicidad de grado 4. Ningún paciente murió de hepatotoxicidad en el Estudio 3011. En los estudios clínicos de fase 3, los pacientes cuyos valores basales de ALT o AST estaban elevados tenían más probabilidades de experimentar elevaciones en la prueba de función hepática que aquellos con valores iniciales normales. Cuando se observaron elevaciones de ALT o AST > 5 X LSN, o elevaciones de bilirrubina > 3 X LSN, ZYTIGA® se suspendió o se discontinuó. En dos casos ocurrió incrementos significativos en las pruebas de función hepática (ver sección Advertencias y precauciones - Hepatotoxicidad e Insuficiencia hepática). Estos dos pacientes con función hepática normal basal experimentaron elevaciones de ALT o AST de 15 a 40 X LSN y elevaciones de bilirrubina de 2 a 6 X LSN. Después de la discontinuación de ZYTIGA®, ambos pacientes presentaron normalización de sus pruebas de función hepática y un paciente fue nuevamente tratado con ZYTIGA® sin recurrencia de las elevaciones. En el Estudio 302, se observaron elevaciones de ALT o AST de grado 3 o 4 en 35 (6.5%) pacientes tratados con ZYTIGA®. Las elevaciones de la aminotransferasa se resolvieron en todos los pacientes excepto en 3 (2 con nueva metástasis hepática múltiple y 1 con elevación de AST aproximadamente 3 semanas después de la última dosis de ZYTIGA®). En los estudios clínicos de fase 3, se reportaron la discontinuación del tratamiento debido al incremento de ALT y AST o función hepática anormal en el 1.1 % de los pacientes tratados con ZYTIGA® y en el 0.6% de los pacientes tratados con el placebo; no se reportaron muertes debido a los eventos de hepatotoxicidad.
En los estudios clínicos, se mitigó el riesgo de hepatotoxicidad excluyendo a los pacientes con hepatitis basal o alteraciones significativas en las pruebas de función hepática. En el estudio 3011, se excluyeron a los pacientes con valores basales de ALT y AST > 2.5 X LSN, bilirrubina > 1.5 X LSN o aquellos con hepatitis viral activa o sintomática o con enfermedad hepática crónica; ascitis o trastornos hemorrágicos secundarios a la insuficiencia hepática. En el estudio 301, se excluyeron a los pacientes con valores basales de ALT y AST ≥2.5 X LSN en ausencia de metástasis hepática y > 5 X LSN en presencia de metástasis hepática. En el estudio 302, los pacientes con metástasis hepática no fueron elegidos y se excluyeron a los pacientes con valores basales de ALT y AST ≥ 2.5 X LSN. Las pruebas de función hepática anormal realizadas en los pacientes que participaron en los estudios clínicos fueron manejadas estrictamente requiriendo la interrupción del tratamiento y permitiendo reanudar el tratamiento solo después que las pruebas de función hepática retornaran a los valores basales del paciente (ver sección Posología y administración - Insuficiencia hepática). Los pacientes con elevaciones de ALT o AST > 20 X LSN no fueron tratados nuevamente. Se desconoce la seguridad de reanudar del tratamiento en dichos pacientes. No se ha comprendido el mecanismo de la hepatotoxicidad asociado con ZYTIGA®.
Experiencia posterior a la comercialización: A continuación, se describen las reacciones adversas identificadas durante la experiencia posterior a la comercialización en base a reportes espontáneos con ZYTIGA®. Las frecuencias se proporcionan de acuerdo a la siguiente convención:
Poco frecuente ≥ 1/1000 y < 1/100, Raro ≥ 1/10000 y < 1/1000
Sistema de clasificación de órganos: Trastornos respiratorios, torácicos y mediastínicos.
Raro: Alveolitis alérgica.
Sistema de clasificación de órganos: Trastornos musculoesqueléticos y del tejido conectivo.
Poco frecuente: Rabdomiolisis, miopatía.
Sistema de clasificación de órganos: Trastornos hepatobiliares.
Raro: Hepatitis fulminante, insuficiencia hepática aguda
INTERACCIONES:
Efecto de los alimentos sobre acetato de abiraterona: La administración de ZYTIGA®con los alimentos incrementa significativamente la absorción de acetato de abiraterona. No se ha establecido la eficacia y la seguridad de ZYTIGA® cuando se administra con los alimentos. ZYTIGA® no se debe tomar con los alimentos (ver sección Posología y administración y Propiedades farmacocinéticas - Absorción).
Interacciones con otros fármacos:
Potencial de otros fármacos que afectan las exposiciones a abiraterona: En un estudio clínico de interacción farmacocinética de sujetos sanos previamente tratados con un inductor potente del CYP3A4 (rifampicina, 600 mg una vez al día durante 6 días) seguido de una dosis única de acetato de abiraterona de 1000 mg, la media plasmática de la AUC∞ de abiraterona se redujo en un 55%.
Los inductores potentes de la CYP3A4 (por ejemplo, fenitoína, carbamazepina, rifampicina, rifabutina, rifapentina, fenobarbital) durante el tratamiento con ZYTIGA® se deben evitar o usar con una cuidadosa evaluación de la eficacia clínica.
En un estudio separado de interacción farmacocinética clínica de sujetos sanos, co-administración de ketoconazol, un inhibidor potente del CYP3A4, no hubo efecto clínicamente significativo sobre la farmacocinética de abiraterona.
Potencial de ZYTIGA® para afectar las exposiciones a otros fármacos: La abiraterona es un inhibidor de las enzimas hepáticas metabolizadoras de fármacos CYP2D6 y CYP2C8. En un estudio clínico para determinar los efectos de acetato de abiraterona (más prednisona) sobre una única dosis de dextrometorfano, sustrato del CYP2D6, la exposición sistémica (AUC) de dextrometorfano se incrementó en aproximadamente 200%. La AUC24 para dextrorfano, el metabolito activo del dextrometorfano, se incrementó aproximadamente en 33%.
Se recomienda precaución cuando ZYTIGA® se administra con fármacos activados o metabolizados por CYP2D6, particularmente con fármacos que tienen un índice terapéutico estrecho. Se debe considerar la reducción de la dosis de los fármacos con índice terapéutico estrecho metabolizados por CYP2D6.
En el mismo estudio para determinar los efectos del acetato de abiraterona (más prednisona) sobre una única dosis de teofilina, sustrato del CYP1A2, no se observó incremento de la exposición sistémica de teofilina.
En un estudio de interacción fármaco-fármaco del CYP2C8 en sujetos sanos, la AUC de pioglitazona se incrementó en un 46% y la AUC para M-III y M-IV, los metabolitos activos de la pioglitazona, disminuyeron un 10% cada uno, cuando pioglitazona se administró junto con una dosis única de 1000 mg de acetato de abiraterona. Aunque estos resultados indican que no se esperan incrementos clínicamente significativos en la exposición cuando ZYTIGA® se combina con fármacos que se eliminan predominantemente por CYP2C8, se debe monitorear en los pacientes signos de toxicidad relacionados con un sustrato del CYP2C8 con un índice terapéutico estrecho si se utiliza concomitantemente con ZYTIGA®.
ESTUDIOS CLÍNICOS:
La eficacia de ZYTIGA® se estableció en tres estudios clínicos de fase 3, aleatorizados, multicéntricos y controlados con placebo (Estudios 3011, 302 y 301) de pacientes con cáncer de próstata metastásico sin tratamiento hormonal previo y cáncer de próstata metastásico resistente a la castración.
El Estudio 3011 enroló pacientes que fueron recientemente diagnosticados (dentro de 3 meses de la aleatorización) con mHNPC que presentaron factores de pronóstico de alto riesgo. El pronóstico de alto riesgo se definió con la presencia de al menos 2 de los siguientes 3 factores de riesgo: (1) escala de Gleason de ≥ 8; (2) presencia de 3 o más lesiones en imágenes óseas; (3) presencia de metástasis visceral medible (excluyendo enfermedad de los ganglios linfáticos). En el grupo activo, se administró ZYTIGA® a una dosis de 1000 mg diarios en combinación con bajas dosis de prednisona o 5 mg de prednisolona una vez al día además del ADT (agonista de LHRH u orquiectomía), lo cual fue el estándar de tratamiento. Los pacientes en el grupo de control recibieron ADT y placebos para ZYTIGA® y prednisona.
El Estudio 302 enroló a pacientes que fueron asintomáticos o levemente sintomáticos y que no han recibido quimioterapia previa, mientras que el Estudio 301 enroló a pacientes que han recibido quimioterapia previa conteniendo un taxano. En ambos estudios, los pacientes estaban utilizando un agonista de LHRH o fueron tratados previamente con orquiectomía. En el grupo de tratamiento activo, se administró ZYTIGA® a una dosis diaria de 1000 mg en combinación con dosis bajas de prednisona o 5 mg de prednisolona a dos veces al día. Los pacientes control recibieron el placebo y dosis bajas de prednisona o 5 mg prednisolona de dos veces al día.
Debido a que los cambios en la concentración sérica del PSA no siempre predicen el beneficio clínico, en todos los estudios los pacientes se mantuvieron con ZYTIGA® hasta que el criterio de discontinuación se cumpliera según lo especificado a continuación para cada estudio.
Estudio 3011 (pacientes con cáncer de próstata metastásico de alto riesgo sin tratamiento hormonal previo (mHNPC) recientemente diagnosticado o cáncer de próstata sensible a hormonas (mHSPC): En el Estudio 3011, (n=1199), la mediana de la edad de los pacientes enrolados fue 67 años. El estado funcional Eastern Cooperative Oncology Group (ECOG, por sus siglas en inglés) fue 0 o 1 en el 97% de los pacientes. Los pacientes con hipertensión no controlada, enfermedad cardiaca significativa, o insuficiencia cardiaca de Clase II o más severa de la NYHA fueron excluidos. Los criterios de valoración co-primarios de la eficacia fueron supervivencia global (SG) y supervivencia libre de progresión radiográfica (SLPr). La mediana de la escala del valor basal del dolor, medido según el Formato corto del cuestionario breve de dolor (BPI-SF, por sus siglas en inglés) fue 2.0 en los grupos de tratamiento y el placebo. Adicionalmente a las mediciones del criterio de valoración co-principal, también se evaluó el beneficio utilizando el tiempo hasta el evento relacionado con el esqueleto (SRE, por sus siglas en inglés), el tiempo hasta terapia subsecuente para el cáncer de próstata, el tiempo hasta el inicio de quimioterapia, el tiempo hasta la progresión del dolor y el tiempo hasta la progresión del PSA.
En el estudio 3011, el tratamiento continuó hasta la progresión de la enfermedad, el retiro de la autorización, la ocurrencia de toxicidad inaceptable o la muerte.
La supervivencia libre de progresión radiográfica se definió como el tiempo desde aleatorización hasta la ocurrencia de la progresión radiográfica o la muerte por cualquier causa. La progresión radiográfica incluyó progresión mediante imágenes óseas (de acuerdo al PCWG2 modificado) o la progresión de lesiones del tejido blando por CT o MRI (de acuerdo con RECIST 1.1).
En el análisis de la SLPr planeado hubo 593 eventos; 239 (40.0%) de los pacientes tratados con ZYTIGA® y 354 (58.8%) de los pacientes tratados con el placebo presentaron evidencia radiográfica de progresión o habían muerto. Se observó una diferencia importante en la SLPr entre los grupos de tratamiento (ver Tabla 2 y Figura 1).
|
Tabla 2: Supervivencia libre de progresión radiográfica-análisis estratificado; población con intención de tratamiento (Estudio PCR3011) |
||
|
AA-P |
Placebo |
|
|
Sujetos aleatorizados |
597 |
602 |
|
Evento |
239 (40.0%) |
354 (58.8%) |
|
Censurados |
358 (60.0%) |
248 (41.2%) |
|
Tiempo hasta el evento (meses) |
||
|
25o percentil (IC del 95%) |
14.59 (11.47, 15.61) |
7.43 (7.29, 10.58) |
|
Mediana (IC del 95%) |
33.02 (29.57, NE) |
14.78 (14.69, 18.27) |
|
75o percentil (IC del 95%) |
NE (NE, NE) |
30.36 (29.24, 39.95) |
|
Rango |
(0.0+, 41.0+) |
(0.0+, 40.6+) |
|
Tasa de 6 meses libres de eventos (IC del 95%) |
0.941 (0.918, 0.957) |
0.867 (0.836, 0.892) |
|
Tasa de 12 meses libres de eventos (IC del 95%) |
0.779 (0.742, 0.812) |
0.611 (0.567, 0.652) |
|
Tasa de 18 meses libres de eventos (IC del 95%) |
0.702 (0.661, 0.739) |
0.476 (0.431, 0.520) |
|
Tasa de 24 meses libres de eventos (IC del 95%) |
0.611 (0.568, 0.652) |
0.347 (0.303, 0.391) |
|
Tasa de 30 meses libres de eventos (IC del 95%) |
0.532 (0.483, 0.579) |
0.250 (0.206, 0.296) |
|
Tasa de 36 meses libres de eventos (IC del 95%) |
0.471 (0.414, 0.526) |
0.209 (0.162, 0.260) |
|
Valor pa |
< 0.0001 |
|
|
Cociente de riesgo (IC del 95%)b |
0.466 (0.394, 0.550) |
|
|
Nota: + = observación censurada, NE=no estimable. La progresión radiográfica y la muerte se consideran al definir el evento SLPr. AA-P= sujetos que recibieron acetato de abiraterona y prednisona. a El valor p es de una prueba de rango logarítmico estratificada por la escala ECOG PS (0/1 o 2) y visceral (ausente o presente). b El cociente de riesgo es de un modelo estratificado proporcional de riesgos. El cociente de riesgo <1 favorece AA-P. |
||
Figura 1: Gráfico Kaplan-Meier de supervivencia libre de progresión radiográfica; población con intención de tratamiento (Estudio PCR3011)
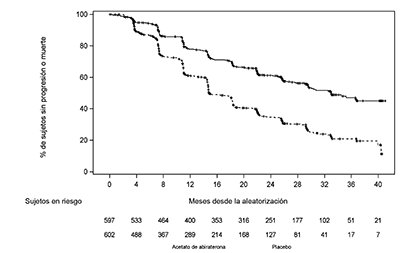
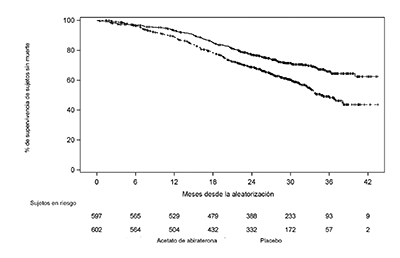
En el primer análisis interino planeado (IA-1) de supervivencia global, habían ocurrido cuatrocientas seis muertes (406; 47.7% del número total de muertes requerido en el análisis final) (169 sujetos en el grupo AA-P y 237 sujetos en el grupo del placebo). Se observó una mejora estadísticamente significativa en la SG a favor de AA-P más ADT con una reducción del 38% en el riesgo de muerte (HR=0.621; IC del 95%: 0.509, 0.756) comparado con el placebo más ADT. La mediana de la supervivencia no se alcanzó en el grupo AA-P frente al 34.7 meses en el grupo del placebo (p<0.0001, cruzando el límite pre-especificado de 0.010 para la SG en el análisis interino 1) (ver Tabla 3 y Figura 2). El estudio no fue ciego basado en la magnitud del beneficio clínico observado y a los pacientes en el grupo del placebo se les ofreció tratamiento con ZYTIGA®. La supervivencia siguió siendo monitoreada después de este IA.
|
Tabla 3: Supervivencia global, análisis estratificado; población con intención de tratamiento (Estudio PCR3011) |
||
|
AA-P |
Placebo |
|
|
Sujetos aleatorizados |
597 |
602 |
|
Evento |
169 (28.3%) |
237 (39.4%) |
|
Censurado |
428 (71.7%) |
365 (60.6%) |
|
Supervivencia global (meses) |
||
|
25o percentil (IC del 95%) |
26.12 (22.74, 30.13) |
19.75 (17.91, 21.82) |
|
Mediana (IC del 95%) |
NE (NE, NE) |
34.73 (33.05, NE) |
|
75o percentil (IC del 95%) |
NE (NE, NE) |
NE (NE, NE) |
|
Rango |
(0.1, 43.5+) |
(1.4+, 43.5+) |
|
Tasa de 12 meses libres de eventos (IC del 95%) |
0.931 (0.908, 0.949) |
0.892 (0.863, 0.914) |
|
Tasa de 24 meses libres de eventos (IC del 95%) |
0.769 (0.732, 0.802) |
0.686 (0.646, 0.723) |
|
Tasa de 36 meses libres de eventos (IC del 95%) |
0.658 (0.608, 0.704) |
0.492 (0.436, 0.546) |
|
Valor pa |
< 0.0001 |
|
|
Cociente de riesgo (IC del 95%)b |
0.621 (0.509, 0.756) |
|
|
Nota: += observación censurada, NE = no estimable. AA-P= sujetos que recibieron acetato de abiraterona y prednisona. a El valor p es de la prueba estratificada de rango logarítmico según la escala ECOG PS (0/1 o 2) y visceral (ausente o presente). b El cociente de riesgo es de un modelo estratificado proporcional de riesgos. El cociente de riesgo <1 favorece a AA-P. |
||
Figura 2: Gráfico Kaplan-Meier de supervivencia global; población con intención de tratamiento (Estudio PCR3011)
Los análisis de subgrupo consistentemente favorecen al tratamiento con ZYTIGA® (ver Figura 3).
|
Figura 3: Supervivencia global por subgrupo; población con intención de tratamiento (Estudio PCR3011) |
||||||
|
GEFOS01F: Supervivencia global por subgrupo Grupos de tratamiento para todos los sujetos y subgrupos Población con intención de tratamiento: (Estudio 212082PCR3011) |
||||||
|
Mediana (meses) |
Eventos/N |
|||||
|
Variable |
Subgrupo |
AA-P |
Placebo |
HR IC del 95% |
AA-P |
Placebo |

Además de las mejorías observadas en la supervivencia global y la SLPr, se demostró el beneficio para ZYTIGA® frente al tratamiento con el placebo en todas las mediciones del criterio de valoración secundario definidos prospectivamente de la siguiente manera:
Tiempo hasta el evento relacionado con el esqueleto (SRE): Hubo una reducción del 30% en el riesgo de eventos relacionados con el esqueleto (HR=0.703; IC del 95%: [0.539, 0.916] p <0.0086). La mediana del tiempo hasta el SRE no se ha alcanzado para el grupo de estudio con ZYTIGA® o el placebo.
Tiempo hasta la progresión del PSA basada en el criterio PCWG2: La mediana del tiempo hasta la progresión del PSA fue 33.2 meses para pacientes que recibieron ZYTIGA® y 7.4 meses para pacientes que recibieron el placebo (HR=0.299; IC del 95%: [0.255, 0.352], p <0.0001).
Tiempo hasta la terapia subsecuente: La mediana del tiempo hasta la terapia subsecuente en el momento del análisis interino no se alcanzó para pacientes que recibieron ZYTIGA® y fue 21.6 meses para los pacientes que recibieron el placebo (HR = 0.415; IC del 95%: [0.346, 0.497], p <0.0001).
Tiempo hasta el inicio de la quimioterapia: La mediana del tiempo hasta el inicio de la quimioterapia no se alcanzó para pacientes que recibieron ZYTIGA® y fue 38.9 meses para los pacientes que recibieron el placebo (HR = 0.443; IC del 95%: [0.349, 0.561], p < 0.0001).
Tiempo hasta la progresión del dolor: La mediana de tiempo hasta la progresión del dolor no se alcanzó para pacientes que recibieron ZYTIGA® y fue 16.6 meses para los pacientes que recibieron el placebo (HR = 0.695; IC del 95%: [0.583, 0.829], p = <0.0001).
La mayoría de los criterios de valoración exploratorios favorecieron al tratamiento con acetato de abiraterona y prednisona (AA-P) sobre el placebo. Una mejoría estadísticamente significativa en la SG específica para cáncer de próstata se observó en el tratamiento AA-P comparado con el placebo (HR=0.547, p<0.0001). Una respuesta confirmada del PSA se observó en el 91.0% de los sujetos en el grupo AA-P y en el 66.8% de los sujetos en el grupo del placebo (riesgo relativo = 1.362; p<0.0001). La tasa de respuesta global (respuesta completa más parcial) en sujetos con enfermedad medida en la línea base fue significativamente mayor en el grupo AA-P comparado con aquellos en el grupo del placebo (p=0.0002).
Los análisis del tiempo hasta la degradación de las medidas del resultado reportado por el paciente (RPP) demostraron consistentemente que el tratamiento con AA-P retrasó la degradación y la progresión del dolor, del estado funcional, de la fatiga y de la calidad de vida asociada con la salud. Basado en el cambio desde la línea base utilizando un modelo de medidas repetidas de efecto mixto, se observaron diferencias estadísticamente significativas entre AA-P y el placebo desde el Ciclo 2 y se mantuvieron a lo largo del estudio.
Estudio 302 (pacientes asintomáticos o levemente sintomáticos que no recibieron quimioterapia previa): En el Estudio 302, (n=1088) la mediana de la edad de los pacientes enrolados fue 71 años para los pacientes tratados con ZYTIGA® más prednisona o prednisolona y 70 años para los pacientes tratados con el placebo más prednisona o prednisolona. El estado funcional ECOG fue 0 para el 76% de los pacientes y 1 para el 24% de los pacientes en ambos grupos. Los pacientes con metástasis visceral fueron excluidos. Los criterios de valoración co-primarios de la eficacia fueron la supervivencia global y la supervivencia libre de progresión radiográfica (SLPr). La valoración basal del dolor fue 0-1 (asintomático) en el 66% de los pacientes y 2-3 (levemente sintomático) en el 26% de los pacientes según lo definido por el Formato corto del cuestionario breve de dolor - (peor dolor en las últimas 24 horas). Además de las mediciones del criterio de valoración co-primario, también se evaluó el beneficio utilizando el tiempo hasta el uso de opiáceos para el dolor oncológico, el tiempo hasta el inicio de la quimioterapia citotóxica, el tiempo hasta el deterioro en la escala funcional ECOG en ≥ 1 punto y el tiempo hasta la progresión del PSA basado en el criterio del Grupo de Trabajo sobre Cáncer de Próstata-2 (PCWG2).
En el Estudio 302, los tratamientos se descontinuaron al momento de la progresión clínica inequívoca. A discreción del investigador, los tratamientos también pudieron ser discontinuados al momento de la progresión radiográfica confirmada.
La supervivencia libre de progresión radiográfica se evaluó con el uso de estudios de imágenes secuenciales definido por el criterio PCWG2 (para lesiones óseas) y el criterio de evaluación de la respuesta en tumores sólidos (RECIST, por sus siglas en inglés) modificado (para lesiones en tejidos blandos). El análisis de la SLPr utilizó la evaluación radiográfica de la progresión con revisión centralizada.
En los análisis planificados de la SLPr se presentaron 401 eventos; 150 (28%) de los pacientes tratados con ZYTIGA® y 251 (46%) de los pacientes tratados con el placebo tuvieron evidencia radiográfica de progresión o habían muerto. Se observó una diferencia significativa en la SLPr entre los grupos de tratamiento (ver Tabla 4 y Figura 4).
|
Tabla 4: Estudio 302: Supervivencia libre de progresión radiográfica de los pacientes tratados con ZYTIGA® o placebo en combinación con prednisona o prednisolona más agonistas de la LHRH u orquiectomía previa |
||
|
ZYTIGA® (N=546) |
Placebo (N=542) |
|
|
Supervivencia libre de progresión radiográfica (SLPr) |
||
|
Progresión o muerte |
150 (28%) |
251 (46%) |
|
Mediana de la SLPr en meses (IC del 95%) |
No se alcanzó (11.66; NE) |
8.3 (8.12; 8.54) |
|
Valor p* |
<0.0001 |
|
|
Cociente de riesgo ** (IC del 95%) |
0.425 (0.347; 0.522) |
|
|
NE = no estimado * El valor p se deriva de una prueba de rango logarítmico estratificada mediante la escala basal ECOG (0 o 1) **Cociente de riesgo (HR) <1 favorable a ZYTIGA® |
||
Figura 4. Curvas de Kaplan Meier de supervivencia libre de progresión radiográfica en pacientes tratados con ZYTIGA® o con placebo en combinación con prednisona o prednisolona más agonistas de la LHRH u orquiectomía previa
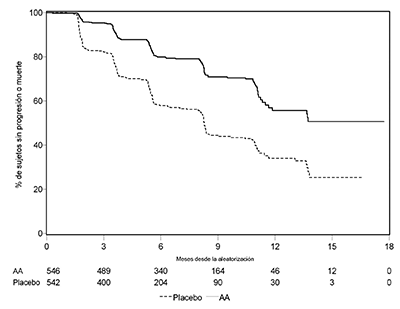
AA=ZYTIGA®
Sin embargo, datos del sujeto continuaron siendo recolectados hasta la fecha del segundo análisis interino de la supervivencia global (SG). La revisión radiográfica del investigador de la SLPr realizado como un análisis de seguimiento de la sensibilidad se presenta en la Tabla 5 y la Figura 5.
Seiscientos siete (607) sujetos tuvieron progresión radiográfica o murieron: 271 (50%) en el grupo de acetato de abiraterona y 336 (62%) en el grupo del placebo. El tratamiento con acetato de abiraterona disminuyó el riesgo de progresión radiográfica o la muerte en 47% comparado con el placebo (HR=0.530; IC del 95%: [0.451, 0.623], p < 0.0001). La mediana de la SLPr fue 16.5 meses en el grupo de acetato de abiraterona y 8.3 meses en el grupo del placebo.
|
Tabla 5: Estudio 302: Supervivencia libre de progresión radiográfica de pacientes tratados ya sea con ZYTIGA® o placebo en combinación con prednisona o prednisolona más análogos de la LHRH u orquiectomía previa (En el segundo análisis interino de la revisión del Investigador de la SG) |
||
|
ZYTIGA® (N=546) |
Placebo (N=542) |
|
|
Supervivencia libre de progresión radiográfica (SLPr) |
||
|
Progresión o muerte |
271 (50%) |
336 (62%) |
|
Mediana de la SLPr en meses (IC del 95%) |
16.5 (13.80, 16.79) |
8.3 (8.05, 9.43) |
|
Valor p* |
< 0.0001 |
|
|
Cociente de riesgo** (IC del 95%) |
0.530 (0.451, 0.623) |
|
|
* Valor p se deriva de una prueba de rango logarítmico estratificado mediante la escala basal ECOG (0 o 1) ** Cociente de riesgo < 1 favorece ZYTIGA® |
||
Figura 5: Curvas de Kaplan Meier de supervivencia libre de progresión radiográfica en pacientes tratados con ZYTIGA® o placebo en combinación con prednisona o prednisolona más análogos de la LHRH u orquiectomía previa (En el segundo análisis interino de la revisión del investigador de la SG)
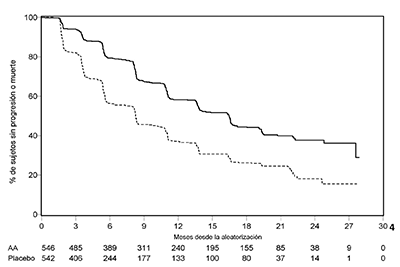
AA=ZYTIGA®
Se realizó un análisis interino (IA) planeado de la supervivencia global después de observar 333 muertes. Se procedió a la apertura del ciego del estudio basado en la magnitud del beneficio clínico observado y a los pacientes en el grupo del placebo se les ofreció tratamiento con ZYTIGA®. La supervivencia global fue mayor para ZYTIGA® que el placebo con una reducción del 25% en el riesgo de muerte (HR = 0.752; IC del 95%: [0.606, 0.934], p = 0.0097), pero la SG no estaba madura y los resultados interinos no cumplieron con el límite de detección pre-especificado para la significación estadística (ver Tabla 6). La supervivencia continuó siendo monitoreada después de este IA.
El análisis final planeado para la SG se realizó después que se observaron 741 muertes (mediana de seguimiento de 49 meses). El 65 % (354 de 546) de los pacientes tratados con ZYTIGA®, en comparación con el 71% (387 de 542) de los pacientes tratados con el placebo, habían muerto. Se demostró un beneficio estadísticamente significativo de la SG a favor del grupo tratado con ZYTIGA® con una reducción del 19.4% en el riesgo de muerte (HR = 0.806; IC del 95%: [0.697, 0.931]; p = 0.0033) y una mejoría en la mediana de la SG de 4.4 Meses (ZYTIGA® 34.7 meses, placebo 30.3 meses) (ver Tabla 6 y Figura 6). Esta mejoría se demostró a pesar que la terapia subsiguiente era común, independientemente de si los pacientes recibieron inicialmente acetato de abiraterona o el placebo. Las terapias subsiguientes en los grupos de pacientes con acetato de abiraterona y el placebo incluyeron acetato de abiraterona, 69 (13%) y 238 (44%); docetaxel, 311 (57%) y 331 (61%); cabazitaxel, 100 (18%) y 105 (19%); y enzalutamida 87 (16%) y 54 (10%) pacientes, respectivamente.
|
Tabla 6: Estudio 302: Supervivencia global de pacientes tratados con ZYTIGA® o el placebo en combinación con prednisona o prednisolona más agonistas de la LHRH u orquiectomía previa |
||
|
ZYTIGA® (N=546) |
Placebo (N=542) |
|
|
Análisis interino de supervivencia |
||
|
Muertes |
147 (27%) |
186 (34%) |
|
Mediana de la supervivencia global en meses (IC del 95%) |
No alcanzado (NE, NE) |
27.2 (25.95, NE) |
|
Valor p* |
0.0097 |
|
|
Cociente de riesgo** (IC del 95%) |
0.752 (0.606, 0.934) |
|
|
Análisis final de supervivencia |
||
|
Muertes |
354 (65%) |
387 (71%) |
|
Mediana de la supervivencia global en meses (IC del 95%) |
34.7 (32.7, 36.8) |
30.3 (28.7, 33.3) |
|
Valor p* |
0.0033 |
|
|
Cociente de riesgo** (IC del 95%) |
0.806 (0.697, 0.931) |
|
|
NE= No estimado * El valor de p se deriva de una prueba de rango logarítmico estratificada mediante la escala basal ECOG (0 o 1) ** Cociente de riesgo < 1 favorece a ZYTIGA®. |
||
Figura 6-Curvas de Kaplan Meier de la supervivencia en pacientes tratados con ZYTIGA® o placebo en combinación con prednisona o prednisolona más agonistas de la LHRH u orquiectomía previa, análisis final
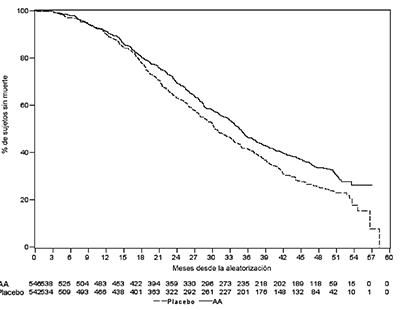
AA=ZYTIGA®
Los análisis de subgrupo consistentemente favorecen al tratamiento con ZYTIGA® (ver Figura 7).
Figura 7: Supervivencia global por subgrupo: Cociente de riesgo e intervalo de confianza del 95%
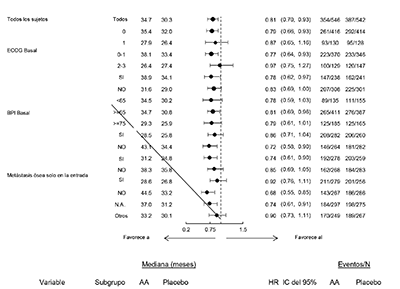
AA = ZYTIGA®; ALK-P = fosfatasa alcalina; BPI = Cuestionario breve del dolor; IC = Intervalo de confianza; ECOG = escala funcional del Eastern Cooperative Oncology Group; HR = Cociente de riesgo; LDH = deshidrogenasa láctica; N.A. = Norteamérica; NE = No evaluable.
Adicionalmente a las mejorías observadas en la supervivencia global y la SLPr, se demostró el beneficio del tratamiento con ZYTIGA® frente al placebo en todas las mediciones del criterio de valoración secundario definido prospectivamente:
Tiempo hasta la progresión del PSA basado en el criterio PCWG2: La mediana de tiempo hasta la progresión del PSA fue 11.1 meses para los pacientes que recibieron ZYTIGA® y 5.6 meses para los pacientes que recibieron placebo (HR = 0.488; IC: del 95%: [0.420, 0.568], p < 0.0001). El tiempo hasta la progresión del PSA aproximadamente se duplicó con el tratamiento con ZYTIGA® (HR = 0.488). La proporción de sujetos con una respuesta confirmada del PSA fue mayor en el grupo de ZYTIGA® que en el grupo del placebo (62% frente al 24%; p < 0.0001).
Tiempo hasta el uso de opiáceos para dolor oncológico: La mediana del tiempo hasta el uso de opiáceos para el dolor por cáncer de próstata al momento del análisis final fue 33.4 para los pacientes que recibieron ZYTIGA® y 23.4 meses para los pacientes que recibieron el placebo (HR = 0.721; IC del 95%: [0.614, 0.846], p < 0.0001).
Tiempo hasta el inicio de la quimioterapia citotóxica: La mediana de tiempo hasta el inicio de la quimioterapia citotóxica fue 25.2 meses para los pacientes que recibieron ZYTIGA® y 16.8 meses para los pacientes que recibieron el placebo (HR = 0.580; IC del 95%: [0.487, 0.691], p < 0.0001).
Tiempo hasta el deterioro en la escala funcional ECOG en ≥ 1 punto: La mediana de tiempo hasta el deterioro en la escala funcional ECOG en ≥ 1 punto fue 12.3 meses para los pacientes que recibieron ZYTIGA® y 10.9 meses para los pacientes que recibieron el placebo (HR = 0.821; IC del 95%: [0.714, 0.943], p = 0.0053).
Los siguientes criterios de valoración del estudio demostraron una ventaja estadísticamente a favor del tratamiento con ZYTIGA®:
Respuesta objetiva: La respuesta objetiva se definió como la proporción de sujetos con una enfermedad medible que alcanzaron una respuesta completa o parcial de acuerdo al criterio RECIST (se requirió que el tamaño basal del ganglio linfático sea ≥ 2 cm para ser considerado una lesión objetivo). La proporción de sujetos con enfermedad medible al momento basal que presentaron una respuesta objetiva fue 36% en el grupo de ZYTIGA® y 16% en el grupo del placebo (p<0.0001).
Dolor: el tratamiento con ZYTIGA® redujo significativamente el riesgo de la progresión de la intensidad del dolor promedio en un 18% en comparación con el placebo (p=0.0490). La mediana de tiempo hasta la progresión fue 26.7 meses en el grupo de ZYTIGA® y 18.4 meses en el grupo del placebo.
Tiempo hasta el deterioro en la escala FACT-P (puntuación total): El tratamiento con ZYTIGA® disminuyó el riesgo del deterioro en la escala FACT-P (puntuación total) en un 22% en comparación con el placebo (p=0.0028). La mediana del tiempo hasta el deterioro en la escala FACT-P (puntuación total) fue 12.7 meses en el grupo de ZYTIGA® y 8.3 meses en el grupo del placebo.
Estudio 301 (pacientes que han recibido quimioterapia previa)
El 11% de los pacientes enrolados en el Estudio 301 tuvieron una puntuación funcional ECOG de 2; el 70% presentó evidencia radiográfica de la progresión de la enfermedad con o sin progresión del PSA; el 70% había recibido una quimioterapia citotóxica previa y el 30% había recibido dos. Se presentó metástasis hepática en el 11% de los pacientes tratados con ZYTIGA®.
Se recomendó que los pacientes siguieran con sus fármacos de estudio hasta que hubo progresión del PSA (incremento confirmado del 25% por encima del punto de valor basal/mínimo del paciente) junto con la progresión radiográfica definida en el protocolo y progresión sintomática o clínica. El criterio de valoración primario de la eficacia fue la supervivencia global.
En un análisis planeado, realizado después de observarse 552 muertes, el 42% (333 de 797) de los pacientes tratados con ZYTIGA®, en comparación con el 55 % (219 de 398) de los pacientes tratados con el placebo, habían muerto. En los pacientes tratados con ZYTIGA® se observó una mejoría estadísticamente significativa en la mediana de la supervivencia global (ver tabla 7 y la figura 8). Se realizó un análisis actualizado de la supervivencia cuando se observaron 775 muertes (97% del número de muertes planeadas para el análisis final). Los resultados a partir de este análisis actualizado de la supervivencia fueron consistentes con los del análisis primario de la supervivencia (ver la tabla 4).
|
Tabla 9. Estudio 301: Supervivencia global de los pacientes tratados con ZYTIGA® o placebo en combinación con prednisona o prednisolona más agonistas de la LHRH u orquiectomía previa |
||
|
ZYTIGA® (N = 797) |
Placebo (N = 398) |
|
|
Analisis primario de la supervivencia |
||
|
Muertes |
333 (42%) |
219 (55%) |
|
Mediana de la supervivencia global en meses (IC del 95%) |
14.8 (14.1; 15.4) |
10.9 (10.2; 12.0) |
|
Valor p |
< 0.0001 |
|
|
Cociente de riesgo *(95% IC) |
0.646 (0.543, 0.768) |
|
|
Análisis actualizado de la supervivencia |
||
|
Muertes |
501 (63%) |
274 (69%) |
|
Mediana de la supervivencia global en meses (IC del 95%) |
15.8 (14.8, 17.0) |
11.2 (10.4, 13.1) |
|
Cociente de riesgo *(IC del 95%) |
0.740 (0.638, 0.859) |
|
|
*Cociente de riesgo < 1 favorable a ZYTIGA® |
||
En todos los puntos temporales de evaluación después de los meses iniciales de tratamiento, una proporción mayor de pacientes tratados con ZYTIGA® seguían vivos, en comparación con la proporción de pacientes tratados con el placebo (ver figura 8).
Figura 8: Curvas de Kaplan-Meier de la supervivencia de los pacientes tratados con ZYTIGA® o el placebo en combinación con prednisona o prednisolona más agonistas de la LHRH u orquiectomía previa
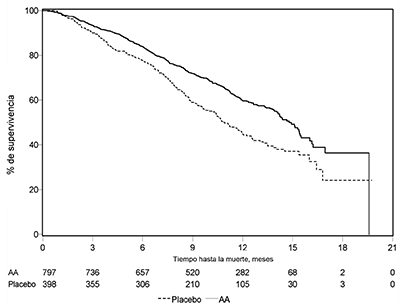
AA=ZYTIGA®
El análisis de la supervivencia de subgrupos mostró un beneficio consistente de la supervivencia para el tratamiento con ZYTIGA® (ver figura 9).
Figura 9. Supervivencia global por subgrupo: Cociente de riesgo e intervalo de confianza del 95%

AA = ZYTIGA®; ALK-P = fosfatasa alcalina; BPI = cuestionario breve del dolor, IC= intervalo de confianza; ECOG = puntuación funcional del Eastern Cooperative Oncology Group; HR = cociente de riesgo; LDH = deshidrogenasa láctica; NA = Norteamérica; NE = no evaluable
Adicionalmente a la mejoría observada en la supervivencia global, todos los criterios de valoración secundarios fueron a favor de ZYTIGA® y fueron estadísticamente significativos después de ajustar para las pruebas tal como se muestra a continuación:
Los pacientes que recibieron ZYTIGA® demostraron un índice de respuesta del PSA total significativamente mayor (definido como una reducción ≥ 50% desde el valor basal), en compáración con los pacientes que recibieron el placebo: un 38% frente al 10%, p < 0.0001.
La mediana del tiempo hasta la progresión del PSA fue 10.2 meses para los pacientes tratados con ZYTIGA® y 6.6 meses para los pacientes tratados con el placebo (HR = 0.580; IC del 95%: [0.462, 0.728], p < 0.0001).
La mediana de la supervivencia libre de progresión radiográfica fue 5.6 meses para los pacientes tratados con ZYTIGA® y 3.6 meses para los pacientes que recibieron el placebo (HR = 0.673; IC del 95%: [0.585, 0.776], p < 0.0001).
Dolor: La proporción de pacientes con alivio del dolor fue, desde el punto de vista estadístico, significativamente mayor en el grupo de ZYTIGA® que en el grupo del placebo (44% frente al 27%, p = 0.0002). Quién respondió al alivio del dolor se definió como un paciente que experimentó al menos una reducción del 30% desde el valor basal en la puntuación BPI-SF de la intensidad del peor dolor durante las últimas 24 horas sin ningún incremento en la puntuación del uso de analgésicos observado en dos evaluaciones consecutivas separadas por cuatro semanas. Solo los pacientes con una puntuación del dolor basal de ≥ 4 y al menos una puntuación de dolor posterior a la basal fueron analizados (n = 512) para el alivio del dolor.
Una proporción inferior de pacientes tratados con ZYTIGA® presentaron progresión del dolor en comparación con los pacientes que tomaron el placebo a los 6 meses (22% frente al 28%), 12 meses (30% frente al 38%) y 18 meses (35% frente al 46%). Se definió la progresión del dolor como un incremento de ≥ 30% desde el valor basal en la puntuación BPI-SF de la intensidad del peor dolor durante las 24 horas previas sin una disminución en la puntuación del uso de analgésicos observado en dos visitas consecutivas, o un incremento de ≥ 30% en la puntuación del uso de analgésicos observado en dos visitas consecutivas. El tiempo hasta la progresión del dolor en el 25° percentil fue 7.4 meses en el grupo de ZYTIGA® frente al 4.7 meses en el grupo del placebo.
Eventos relacionados con el esqueleto: Una proporción menor de pacientes en el grupo de ZYTIGA® presentó eventos relacionados con el esqueleto en comparación con el grupo del placebo a los 6 meses (18% frente al 28%), 12 meses (30% frente al 40%) y 18 meses (35% frente al 40%). El tiempo hasta el primer evento relacionado con el esqueleto en el 25° percentil en el grupo ZYTIGA® fue el doble que en el grupo de control, con 9.9 meses frente a 4.9 meses. Un evento relacionado con el esqueleto se definió se definió como una fractura patológica, compresiones medular, radioterapia paliativa ósea o cirugía ósea.
INFORMACIÓN NO CLÍNICA:
Carcinogénesis y genotoxicidad: El acetato de abiraterona no fue carcinogénico en un estudio de 6 meses en ratón transgénico (Tg.rasH2). En un estudio de carcinogenicidad de 24 meses en la rata, el acetato de abiraterona incrementó la incidencia de neoplasmas celulares intersticiales en los testículos. Este hallazgo se considera relacionado con la acción farmacológica de la abiraterona y la rata específica. El acetato de abiraterona no fue carcinogénico en ratas hembra.
El acetato de abiraterona y la abiraterona carecieron de potencial genotóxico en el panel estándar de pruebas de genotoxicidad, incluyendo un ensayo in vitro de mutación inversa bacteriana (la prueba de Ames), una prueba in vitro de aberraciones cromosómicas en mamíferos (utilizando linfocitos humanos) y un ensayo in vitro de micronúcleos de rata.
Toxicología reproductiva: En estudios de fertilidad en ratas macho y hembra, el acetato de abiraterona redujo la fertilidad, la cual completamente reversible en 4 a 16 semanas después de la suspensión del acetato de abiraterona.
En un estudio de toxicidad para el desarrollo en la rata, el acetato de abiraterona afectó el embarazo incluyendo la reducción del peso fetal y la supervivencia. Se observaron efectos sobre los genitales externos, si bien acetato de abiraterona no es teratogénico.
En estos estudios de fertilidad y toxicidad para el desarrollo realizados en la rata, todos los efectos estuvieron relacionados con la actividad farmacológica de la abiraterona.
ZYTIGA® está contraindicado en el embarazo (ver sección Contraindicaciones - Embarazo y Embarazo, lactancia y fertilidad- Embarazo).
Toxicología animal: En todos los estudios de toxicidad animal, los niveles de testosterona circulante fueron reducidas significativamente. Como resultado, se observaron reducción del peso de los órganos y cambios morfológicos y/o histopatológicos en los órganos reproductivos, las glándulas suprarrenales, pituitaria y mamaria. Todos los cambios mostraron reversibilidad completa o parcial. Los cambios en los órganos reproductivos y los órganos sensibles a los andrógenos son consistentes con la farmacología de la abiraterona. Todos los cambios hormonales relacionados con el tratamiento se revirtieron o se demostró que se resolvieron después de un período de recuperación de 4 semanas.
Después de un tratamiento crónico a partir de las 13 semanas, se observó en el hígado de la rata y monos hiperplasia del conducto biliar/ células ovales, asociada con el incremento de los niveles de fosfatasa alcalina y/o bilirrubina sérica. Después de un período de recuperación de 4 semanas, los parámetros séricos se revirtieron, mientras que persistió la hiperplasia del conducto biliar/células ovales.
Se observaron cataratas en ratas después de 26 semanas de tratamiento. Estos cambios seguían presentes después de un período de recuperación de 4 semanas. No se observaron cataratas en monos después de 39 semanas de tratamiento.
ADVERTENCIAS Y PRECAUCIONES:
Hipertensión, hipocalemia y retención de líquidos debido al exceso de mineralocorticoides: ZYTIGA® puede causar hipertensión, hipocalemia y retención de líquidos (ver sección Reacciones adversas) como una consecuencia de los niveles incrementados de los mineralocorticodes como resultado de la inhibición de CYP17 (ver sección Propiedades farmacológicas - Mecanismo de acción). La coadministración de un corticosteroide suprime el efecto de la hormona adrenocorticotropina (ACTH), produciendo una reducción en la incidencia y la severidad de estas reacciones adversas. Se requiere precaución al tratar pacientes cuyas condiciones médicas subyacentes podrian estar comprometidas por el incremento de la presión sanguínea, hipocalemia o retención de líquidos; por ejemplo, aquellos con insuficiencia cardiaca, infarto de miocardio reciente o arri™ia ventricular.
ZYTIGA® se debe utilizar con precaución en pacientes con antecedente de enfermedad cardiovascular. No se ha establecido la seguridad de ZYTIGA® en pacientes con una fracción de eyección ventricular izquierda (FEVI) < 50% o insuficiencia cardiaca de clase III o IV en la escala de la New York Heart Association (NYHA por sus siglas en inglés) (en el Estudio 301) o de clase II a IV en la escala de la NYHA (en los estudios 3011 y 302) (ver sección Reacciones adversas y Propiedades farmacológicas - Estudios clínicos). Antes del tratamiento con ZYTIGA®, se debe controlar la hipertensión y se debe corregir la hipocalemia. La presión sanguínea, el potasio sérico y la retención de líquidos se deben monitorear al menos una vez al mes.
Hepatotoxicidad e insuficiencia hepática: En estudios clínicos controlados ocurrieron incrementos significativos de las enzimas hepáticas que conllevaron a discontinuar el fármaco o a modificar la dosificación (ver sección Reacciones adversas). Se deben medir los niveles de la bilirrubina y de la transaminasa sérica y antes de iniciar el tratamiento con ZYTIGA®, cada dos semanas durante los tres primeros meses de tratamiento, y posteriormente cada mes. Si los signos o síntomas clínicos sugieren el desarrollo de hepatotoxicidad, se deben medir inmediatamente las transaminasas séricas. Si en cualquier momento la ALT o AST se incrementa cinco veces por encima del límite superior de normalidad o si la bilirrubina se incrementa tres veces por encima del límite superior de normalidad, se debe interrumpir de inmediato el tratamiento con ZYTIGA® y se debe monitorear cercanamente la función hepática.
Solo se puede reanudar el tratamiento con ZYTIGA® después que las pruebas de la función hepática retornen a los valores basales del paciente y con un nivel de dosis reducida (ver sección Posología y administración - Insuficiencia hepática).
Si los pacientes desarrollan hepatotoxicidad severa (ALT o AST 20 veces por encima del límite superior de normalidad) en cualquier momento durante la terapia, se debe discontinuar ZYTIGA® y los pacientes no deben ser tratados nuevamente con ZYTIGA®.
No existen datos sobre eficacia y seguridad clínica de dosis múltiples de acetato de abiraterona cuando se administró a pacientes con insuficiencia hepática moderada o severa (Child- Pugh Clase B o C). No se pueden predecir ajustes de dosis. ZYTIGA® se debe usar con precaución en pacientes con insuficiencia hepática moderada solo si el beneficio claramente supera el posible riesgo (ver sección Posología y administración - Insuficiencia hepática y Propiedades farmacocinéticas - Poblaciones especiales). No se debe utilizar ZYTIGA® en pacientes con insuficiencia hepática severa (ver sección Posología y administración - Insuficiencia hepática y Propiedades farmacocinéticas - Poblaciones especiales).
Ha habido reportes raros posteriores a la comercialización de insuficiencia hepática aguda y hepatitis fulminante, algunos con resultado mortal (ver sección Reacciones adversas).
Retiro de corticosteroides y tratamiento de las situaciones de estrés: Se recomienda precaución y se debe monitorear la insuficiencia adrenocortical si los pacientes dejan de tomar prednisona o la prednisolona. Si se continua con ZYTIGA® despues de retirar los corticosteroides, se debe monitorear a los pacientes por síntomas de exceso de mineralocorticoides (ver sección Advertencias y precauciones - Hipertensión, hipocalemia y retención de líquidos debido al exceso de mineralocorticoides).
En pacientes tratados con prednisona o prednisolona que están sometidos a estrés inusual, se puede indicar incrementar la dosificación de un corticosteroide antes, durante y después de la situación estresante.
Uso con quimioterapia: No se ha establecido la seguridad y eficacia del uso concomitante de ZYTIGA® con quimioterapia citotóxica (ver sección Propiedades farmacológicas-Estudios clínicos).
POSOLOGÍA Y ADMINISTRACIÓN:
Posología: La dosis recomendada de ZYTIGA® es 1000 mg (cuatro tabletas de 250 mg) como una dosis única diaria que no se debe tomar con los alimentos. ZYTIGA® se debe tomar al menos dos horas después de comer y no se debe ingerir alimentos por al menos una hora después de tomar ZYTIGA®. Las tabletas se deben ingerir enteras con agua (ver sección Propiedades farmacocinéticas - Absorción).
Dosis de prednisona y prednisolona: Para cáncer de próstata metastásico sin tratamiento hormonal previo (mHNCP) o cáncer de próstata metastásico sensible a hormonas (mHSPC) se utiliza ZYTIGA® con 5 mg diarios de prednisona o prednisolona.
Para cáncer de próstata metastásico resistente a la castración (mCRPC), se utiliza ZYTIGA® con 10 mg diarios de prednisona o prednisolona.
Monitoreo recomendado: Se debe medir la bilirrubina y las transaminasas séricas antes de iniciar el tratamiento con ZYTIGA®, cada dos semanas durante los tres primeros meses de tratamiento y posteriormente una vez al mes. Se debe monitorear mensualmente la presión arterial, el potasio sérico y la retención de líquidos (ver sección Advertencias y precauciones - Hipertensión, hipocalemia y retención de líquidos debido al exceso de mineralocorticoides y Hepatotoxicidad e Insuficiencia hepática).
Insuficiencia hepática: No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve preexistente. No existen datos sobre la eficacia y seguridad clínica de dosis múltiples de acetato de abiraterona cuando se administró a pacientes con insuficiencia hepática moderada o severa (Child- Pugh Clase B o C). No se pueden predecir ajustes de dosis. ZYTIGA® se debe usar con precaución en pacientes con insuficiencia hepática moderada, solo si el beneficio claramente supera el posible riesgo (ver sección Advertencias y precauciones- Hepatotoxicidad e insuficiencia hepática y Propiedades farmacocinéticas - Poblaciones especiales). ZYTIGA® no se debe usar en pacientes con insuficiencia hepática severa (ver sección Advertencias y precauciones - Hepatoxicidad e insuficiencia hepática y Propiedades farmacocinéticas - Poblaciones especiales).
En pacientes que desarrollan hepatotoxicidad durante el tratamiento con ZYTIGA® (incremento de la alanina aminotransferasa (ALT) o aspartato aminotransferasa (AST) 5 veces por encima del límite superior de normalidad, o incremento de la bilirrubina de 3 veces por encima del límite superior de normalidad), se debe suspender el tratamiento inmediatamente hasta que las pruebas de función hepática sean normales (ver sección Advertencias y precauciones - Hepatotoxicidad e insuficiencia hepática). Al reanudar el tratamiento, después que las pruebas de la función hepática hayan retornado a los valores basales del paciente, se puede administrar una dosis reducida de 500 mg (dos tabletas de 250 mg) una vez al día. En los pacientes que estan siendo tratados nuevamente, se debe monitorear la bilirrubina y las transaminasas séricas al menos cada dos semanas durante tres meses y posteriormente una vez al mes. Si vuelve ocurrir hepatotoxicidad con la dosis reducida de 500 mg al día, discontinuar el tratamiento con ZYTIGA®. No se deben tomar las dosis reducidas con los alimentos (ver sección Posología y administración - Posología).
Si los pacientes desarrollan hepatotoxicidad severa (ALT o AST 20 veces por encima del límite superior de normalidad) en cualquier momento durante la terapia, se debe discontinuar ZYTIGA® y los pacientes no deben ser tratados nuevamente con ZYTIGA®.
Insuficiencia renal: No es necesario ajustar la dosis en pacientes con insuficiencia renal (ver sección Propiedades farmacocinéticas - Poblaciones especiales).
INSTRUCCIONES DE USO Y MANIPULACIÓN: Por su mecanismo de acción, ZYTIGA® puede dañar un embrión o feto en desarrollo; por esta razón, las mujeres que estén o puedan estar embarazadas no deben manipular ZYTIGA® sin protección, p. ej., guantes (consulte Embarazo, lactancia y fertilidad-Embarazo).
Cualquier producto no utilizado o material de desecho debe eliminarse de acuerdo con las normas locales.
INSTRUCCIONES DE USO, MANIPULACIÓN Y ELIMINACIÓN: Basado en su mecanismo de acción, ZYTIGA® puede dañar a un feto en desarrollo; por lo tanto, las mujeres que están o puedan estar embarazadas no deben manipular ZYTIGA® 250 mg tabletas no recubiertas sin protección, por ejemplo, guantes (ver sección Embarazo, lactancia y fertilidad - Embarazo).
Cualquier producto no utilizado o material de desecho se debe eliminar de acuerdo con los requerimientos locales.
SOBREDOSIS: La experiencia en humanos de sobredosis con ZYTIGA® es limitada.
No existe antídoto específico. En caso de una sobredosis, se debe interrumpir la administración de ZYTIGA® y se deben considerar medidas generales de soporte, incluyendo el monitoreo de arri™ias. También se debe evaluar la función hepática.
PRESENTACIÓN: ZYTIGA® Tabletas, caja por 1 frasco por 120 tabletas (Reg. San. INVIMA 2012M-0013333).
JANSSEN-CILAG, S. A.
Av. Calle 26 No. 69-76 - Edificio Elemento
Torre 2 - Piso 11 - PBX: (+57) 1 9271200 - Bogotá, D. C.
ALMACENAMIENTO: Almacenar a una temperatura inferior a los 30 °C.Mantener fuera del alcance de los niños.
CONDICIONES DE ALMACENAMIENTO: Almacenar a una temperatura inferior a los 30 °C.
Mantener fuera de la vista y del alcance de los niños.
NATURALEZA Y CONTENIDO DEL ENVASE: ZYTIGA® está disponible en frascos redondeados blancos de polietileno de alta densidad, provistos de un tapón de polipropileno. El envase tiene tamaño para 120 tabletas de 250 mg.


