

DARZALEX
DARATUMUMAB
Concentrado en solución para infusión
Caja, 1 Vial(es), 5 ml, 100 Miligramos
Caja, 1 Vial(es), 20 ml, 400 Miligramos
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA:
Cada mL contiene:
Daratumumab 20 mg
Excipientes c.s.p
Para consultar la lista completa de excipientes, ver Lista de excipientes.
INDICACIONES Y USO:
DARZALEX™ está indicado:
• En combinación con lenalidomida y dexametasona, o bortezomib y dexametasona, para el tratamiento de pacientes con mieloma múltiple que han recibido por lo menos una terapia previa.
• Como monoterapia, para el tratamiento de pacientes con mieloma múltiple que han recibido por lo menos tres líneas previas de terapia incluyendo un inhibidor de proteasoma (IP) y un agente inmunomodulador o que son doble refractarios a un IP y a un agente inmunomodulador.
DATOS FARMACÉUTICOS:
Lista de excipientes: Ácido acético glacial, acetato de sodio trihidrato, cloruro de sodio, manitol, polisorbato 20 y agua para inyección.
Incompatibilidades: Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección Precauciones especiales de eliminación y otras manipulaciones.
Período de validez:
• Viales sin abrir: 18 meses
• Tras la dilución: Desde un punto de vista microbiológico, a menos que el método de apertura/dilución excluya el riesgo de contaminación microbiana, el producto debe usarse inmediatamente. Si no se usa inmediatamente, las condiciones y los tiempos de conservación en uso son responsabilidad del usuario y no deben ser superiores a 24 horas en condiciones de refrigeración (entre 2° C y 8° C) protegido de la luz, seguidas de 15 horas (incluido el tiempo de perfusión) a temperatura ambiente (entre 15°C y 25°C) y con luz ambiente.
Precauciones especiales de conservación:
Almacenar de 2°C y 8°C.
No congelar. No agitar.
Para protegerlo de la luz, mantenga el vial en su envase original.
Manténgase fuera de la vista y del alcance de los niños.
Para las condiciones de conservación tras la dilución del medicamento, ver sección Período de validez.
CONTRAINDICACIONES:
Ninguna.
PRECAUCIONES ESPECIALES PARA LA ELIMINACIÓN Y OTRAS MANIPULACIONES:
Este medicamento es únicamente para un solo uso.
Preparar la solución para perfusión utilizando una técnica aséptica tal y como se indica a continuación:
• Calcular la dosis (mg) y el volumen total (ml) de solución de DARZALEX™ que se precisan y el número de viales necesarios de DARZALEX™ en función del peso del paciente.
• Comprobar que la solución de DARZALEX™ sea entre incolora y amarilla. No usar si presenta partículas opacas, cambios de color o partículas extrañas de otro tipo.
• Utilizando una técnica aséptica, extraer un volumen de cloruro de sodio al 0.9% de la bolsa/envase de perfusión equivalente al volumen necesario de la solución de DARZALEX™.
• Extraer la cantidad necesaria de la solución de DARZALEX™ y diluirla hasta el volumen apropiado añadiéndola a una bolsa/envase de perfusión que contenga cloruro de sodio al 0.9% (ver sección Posología y forma de administración). Las bolsas/envases de perfusión deben ser de polivinilcloruro (PVC), polipropileno (PP), polietileno (PE) o mezcla de poliolefinas (PP + PE). Diluir en condiciones asépticas apropiadas. Desechar la parte sobrante sin usar que quede en el vial.
• Invertir suavemente la bolsa/envase para mezclar la solución. No agitar.
• Antes de la administración, hacer una inspección visual de los medicamentos parenterales para descartar la presencia de partículas sólidas y cambios de color. La solución diluida puede presentar partículas proteínicas muy pequeñas, entre translúcidas y blancas, ya que daratumumab es una proteína. No usar si se observan partículas opacas visibles, cambios de color o partículas extrañas.
• Como DARZALEX™ no contiene ningún conservante, la solución diluida se debe administrar en un plazo de 15 horas (incluido el tiempo de perfusión) a temperatura ambiente (entre 15°C y 25°C) y con luz ambiente.
• Si no se usa inmediatamente, la solución diluida puede conservarse antes de su administración durante un máximo de 24 horas en condiciones de refrigeración (entre 2 °C y 8 °C) y protegida de la luz. No congelar.
• Administrar la solución diluida mediante perfusión intravenosa utilizando para ello un equipo de perfusión con regulador de flujo y filtro incorporado estéril y apirógeno de polietersulfona (PES) con escasa fijación proteínica (tamaño de poro, 0.22 o 0,2 µm). Se deben usar equipos de administración de poliuretano (PU), polibutadieno (PBD), PVC, PP o PE.
• DARZALEX™ no se debe administrar junto con otros fármacos a través de la misma vía intravenosa.
• No se debe conservar y reutilizar ninguna parte sobrante de la solución para perfusión sin usar. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Fecha de revisión del texto: Marzo 2017
Versión del texto de referencia: USPI 21 Noviembre 2016
Aprobado por DIGEMID: 12 – Abril - 2017
Titular del registro sanitario:
JOHNSON & JOHNSON DEL PERÚ S.A.
Av. Canaval y Moreyra N° 480, Int. 901 y 1301
San Isidro-Lima
REACCIONES ADVERSAS:
Las siguientes reacciones adversas graves también se describen en otras partes de la ficha técnica:
• Reacciones a la perfusión (ver sección Advertencia y precauciones).
• Neutropenia (ver sección Advertencia y precauciones).
• Trombocitopenia (ver sección Advertencia y precauciones).
Reacciones adversas en ensayos clínicos: Debido a que los ensayos clínicos son conducidos bajo condiciones muy variables, las tasas de las reacciones adversas en los ensayos clínicos de un fármaco no pueden ser comparados directamente con las tasas en los ensayos clínicos de otro fármaco y no pueden reflejar las tasas observadas en la práctica.
Los datos de seguridad descritos a continuación reflejan la exposición a DARZALEX™ (16 mg/kg) en 717 pacientes con mieloma múltiple incluyendo 526 pacientes a partir de dos ensayos activo controlado de Fase 3 que recibieron DARZALEX™ en combinación con lenalidomida (DRd, n=283; Estudio 3) o bortezomib (DVd, n=243; Estudio 4) y cuatro ensayos clínicos abiertos en los que los pacientes recibieron DARZALEX™ ya sea en combinación con lenalidomida (n=35) o como monoterapia (n=156).
Tratamiento combinado con lenalidomida: Las reacciones adversas descritas en la Tabla 4 reflejan la exposición al DARZALEX™ (grupo DRd) con una mediana de duración del tratamiento de 13.1 meses (rango: 0 a 20.7 meses) y una mediana de duración del tratamiento de 12.3 meses (rango: 0.2 a 20.1 meses) para el grupo de lenalidomida (Rd) en el Estudio 3. Las reacciones adversas más frecuentes (≥20%) fueron reacciones a la perfusión, diarrea, náusea, fatiga, pirexia, infección del tracto respiratorio superior, espasmos musculares, tos y disnea. La incidencia general de las reacciones adversas graves fue 49% para el grupo de DRd en comparación con el 42% para el grupo de Rd. Las reacciones adversas graves con por lo menos una incidencia mayor al 2% en el grupo DRd en comparación con el grupo de Rd fueron neumonía (12% frente al 10% de Rd), infección del tracto respiratorio superior (7% frente a 4% de Rd), influenza y pirexia (3% de DRd frente a 1% de Rd para cada uno).
Las reacciones adversas resultaron en la discontinuación para 7% (n=19) de los pacientes en el grupo de DRd frente al 8% (n=22) en el grupo de Rd.
|
Tabla 4: Reacciones adversas reportadas en ≥ 10% de los pacientes y con por lo menos una frecuencia mayor del 5% en el grupo de DRd en el Estudio 3. |
||||||
|
Reacción Adversa |
DRd (N=283) % |
Rd (N=281)% |
||||
|
Cualquier Grado |
Grado 3 |
Grado 4 |
Cualquier Grado |
Grado 3 |
Grado 4 |
|
|
Reacciones de la perfusióna |
48 |
5 |
0 |
0 |
0 |
0 |
|
Trastornos gastrointestinales |
||||||
|
Diarrea |
43 |
5 |
0 |
25 |
3 |
0 |
|
Náusea |
24 |
1 |
0 |
14 |
0 |
0 |
|
Vómitos |
17 |
1 |
0 |
5 |
1 |
0 |
|
Trastornos generales y condiciones en el sitio de administración |
||||||
|
Fatiga |
35 |
6 |
<1 |
28 |
2 |
0 |
|
Pirexia |
20 |
2 |
0 |
11 |
1 |
0 |
|
Infecciones e infestaciones |
||||||
|
Infección del tracto respiratorio superiorb |
65 |
6 |
<1 |
51 |
4 |
0 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||||||
|
Espasmos musculares |
26 |
1 |
0 |
19 |
2 |
0 |
|
Trastornos del sistema nervioso |
||||||
|
Dolor de cabeza |
13 |
0 |
0 |
7 |
0 |
0 |
|
Trastornos respiratorios , torácicos y mediastínicos |
||||||
|
Tosc |
30 |
0 |
0 |
15 |
0 |
0 |
|
Disnead |
21 |
3 |
<1 |
12 |
1 |
0 |
|
Leyenda: D = daratumumab, Rd = lenlidomida-dexametasona. a La reacción de la perfusión incluye los términos determinados por los investigadores para estar relacionados a la perfusión, ver descripción de reacciones a la perfusión a continuación. b Infección del tracto respiratorio superior, bronquitis, sinusitis, infección viral del tracto respiratorio, rinitis, faringitis, infección del tracto respiratorio, infección por metapneumovirus, traqueobronquitis, infección viral del tracto respiratorio superior, laringitis, infección por virus sincicial respiratorio, faringitis estafilocócica, tonsilitis, faringitis viral, sinusitis aguda, nasofaringitis, bronquiolitis, bronquitis viral, faringitis estreptocócica, traqueítis, infección bacteriana del tracto respiratorio superior, bronquitis bacteriana, epiglotitis, laringitis viral, candidiasis orofaríngea, moniliasis respiratoria, rinitis viral, tonsilitis aguda, infección por rinovirus. c Tos, tos productiva, tos alérgica. d Disnea, disnea por esfuerzo. |
||||||
Las anormalidades de laboratorio que empeoran durante el tratamiento desde el valor basal se enumeran en la Tabla 5.
|
Tabla 5: Anormalidades de laboratorio de hematología emergentes del tratamiento en el Estudio 3 |
||||||
|
DRd (N=283) |
Rd (N=281) |
|||||
|
Cualquier Grado |
Grado 3 |
Grado 4 |
Todos los Grados |
Grado 3 |
Grado 4 |
|
|
Anemia |
52 |
13 |
0 |
57 |
19 |
0 |
|
Trombocitopenia |
73 |
7 |
6 |
67 |
10 |
5 |
|
Neutropenia |
92 |
36 |
17 |
87 |
32 |
8 |
|
Linfopenia |
95 |
42 |
10 |
87 |
32 |
6 |
|
Leyenda: D=Daratumumab, Rd=lenlidomida-dexametasona |
||||||
Tratamiento combinado con bortezomib: Las reacciones adversas descritas en la tabla 6 reflejan la exposición a DARZALEX™ (grupo de DVd) durante una mediana de duración del tratamiento de 6.5 meses (rango: 0 a 14.8 meses) y una mediana de duración del tratamiento de 5.2 meses (rango: 0.2 a 8.0 meses) para el grupo de bortezomib (Vd) en el Estudio 4. Las reacciones adversas más frecuentes (>20%) fueron reacciones a la perfusión, diarrea, edema periférico, infección del tracto respiratorio superior, neuropatía sensitiva periférica, tos y disnea. La incidencia general de las reacciones adversas graves fue 42% para el grupo de DVd en comparación con el 34% para el grupo de Vd. Las reacciones adversas graves con al menos una incidencia mayor al 2% en el grupo de DVd en comparación con el grupo de Vd fueron infección del tracto respiratorio superior (5% de DVd frente a 2% de Vd), diarrea y fibrilación auricular (2% de DVd frente a 0% de Vd para cada uno).
Las reacciones adversas resultaron en discontinuaciones para el 7% (n=18) de los pacientes en el grupo de DVd frente al 9% (n=22) en el grupo de Vd.
|
Tabla 6: Reacciones adversas reportadas en ≥ 10% de los pacientes y al menos una frecuencia mayor al 5% en el grupo de DVd del Estudio 4 |
||||||
|
Reacción adversa |
Dvd (N=243)% |
Vd (N=237)% |
||||
|
Cualquier Grado |
Grado 3 |
Grado 4 |
Cualquier Grado |
Grado 3 |
Grado 4 |
|
|
Reacciones a la perfusióna |
45 |
9 |
0 |
0 |
0 |
0 |
|
Trastornos gastrointestinales |
||||||
|
Diarrea |
32 |
3 |
<1 |
22 |
1 |
0 |
|
Vómitos |
11 |
0 |
0 |
4 |
0 |
0 |
|
Trastornos generales y condiciones en el sitio de administración |
||||||
|
Edema periféricob |
22 |
1 |
0 |
13 |
0 |
0 |
|
Pirexia |
16 |
1 |
0 |
11 |
1 |
0 |
|
Infecciones e infestaciones |
||||||
|
Infección del tracto respiratorio superiorc |
44 |
6 |
0 |
30 |
3 |
<1 |
|
Trastornos del sistema nervioso |
||||||
|
Neuropatía sensitiva periférica |
47 |
5 |
0 |
38 |
6 |
<1 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||||
|
Tosd |
27 |
0 |
0 |
14 |
0 |
0 |
|
Disneae |
21 |
4 |
0 |
11 |
1 |
0 |
|
Leyenda: D=daratumumab, Vd=bortezomib-dexametasona. a La reacción a la perfusión incluye los términos determinados por los investigadores para estar relacionados a la perfusión, ver descripción de reacciones a la perfusión debajo. b Edema periférico, edema, edema generalizado, hinchazón periférica c Infección del tracto respiratorio superior, bronquitis, sinusitis, infección viral del tracto respiratorio, rinitis, faringitis, infección del tracto respiratorio, infección por metapneumovirus, traqueobronquitis, infección viral del tracto respiratorio superior, laringitis, infección por virus sincicial respiratorio, faringitis estafilocócica, tonsilitis, faringitis viral, sinusitis aguda, nasofaringitis, bronquiolitis, bronquitis viral, faringitis estreptocócica, traqueítis, infección bacteriana del tracto respiratorio superior, bronquitis bacteriana, epiglotitis, laringitis viral, candidiasis orofaríngea, moniliasis respiratoria, rinitis viral, tonsilitis aguda, infección por rinovirus. d Tos, tos productiva, tos alérgica e Disnea, disnea por esfuerzo |
||||||
Las anormalidades de laboratorio que empeoran durante el tratamiento están descritas en la Tabla 7.
|
Tabla 7: Anormalidades de laboratorio de hematología emergentes del tratamiento en el Estudio 4. |
||||||
|
DVd (N=243) % |
|
|
Vd (N=237) % |
|
|
|
|
|
Cualquier Grado |
Grado 3 |
Grado 4 |
Cualquier Grado |
Grado 3 |
Grado 4 |
|
Anemia |
48 |
13 |
0 |
56 |
14 |
0 |
|
Trombocitopenia |
90 |
28 |
19 |
85 |
22 |
13 |
|
Neutropenia |
58 |
12 |
3 |
40 |
5 |
<1 |
|
Linfopenia |
89 |
41 |
7 |
81 |
24 |
3 |
|
Leyenda: D=Daratumumab, Vd= bortezomib-dexametasona. |
||||||
Monoterapia: Los datos de seguridad reflejan la exposición a DARZALEX™ en 156 pacientes adultos con mieloma múltiple recidivante y refractario tratados con 16 mg/kg de DARZALEX™ en tres ensayos clínicos abiertos. La mediana de la duración de exposición fue 3.3 meses (rango: 0.03 a 20.04 meses). Las reacciones adversas graves fueron reportadas en 51 pacientes (33%). Las reacciones adversas graves más frecuentes fueron neumonía (6%), deterioro de la salud física general (3%) y pirexia (3%).
Las reacciones adversas produjeron retardo del tratamiento para 24 pacientes (15%), más frecuentemente para las infecciones. Las reacciones adversas produjeron discontinuaciones para 6 pacientes (4%).
Las reacciones adversas que ocurren en por lo menos 10% de los pacientes se presentan en la Tabla 8. La Tabla 9 describe las anormalidades de laboratorio de Grado 3-4 reportadas a una tasa de ≥10%.
|
Tabla 8: Reacciones adversas con incidencia ≥ 10% en pacientes con mieloma múltiple tratado con 16 mg/kg de DARZALEX™ |
|||
|
Reacción adversa |
DARZALEX™ 16 mg/kg N= 156 |
||
|
Incidencia (%) |
|||
|
Cualquier Grado |
Grado 3 |
Grado 4 |
|
|
Reacción a la perfusióna |
48 |
3 |
0 |
|
Trastornos generales y condiciones en el sitio de administración |
|||
|
Fatiga |
39 |
2 |
0 |
|
Pirexia |
21 |
1 |
0 |
|
Escalofríos |
10 |
0 |
0 |
|
Trastornos respiratorios, torácicos y mediastínicos |
|||
|
Tos |
21 |
0 |
0 |
|
Congestión nasal |
17 |
0 |
0 |
|
Disnea |
15 |
1 |
0 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
|||
|
Dolor de espalda |
23 |
2 |
0 |
|
Artralgia |
17 |
0 |
0 |
|
Dolor en extremidad |
15 |
1 |
0 |
|
Dolor torácico musculoesquelético |
12 |
1 |
0 |
|
Infecciones e infestaciones |
|||
|
Infección del tracto respiratorio superior |
20 |
1 |
0 |
|
Nasofaringitis |
15 |
0 |
0 |
|
Neumoníab |
11 |
6 |
0 |
|
Trastornos gastrointestinales |
|||
|
Náusea |
27 |
0 |
0 |
|
Diarrea |
16 |
1 |
0 |
|
Estreñimiento |
15 |
0 |
0 |
|
Vómitos |
14 |
0 |
0 |
|
Trastornos del metabolismo y de la nutrición |
|||
|
Disminución del apetito |
15 |
1 |
0 |
|
Trastornos del sistema nervioso |
|||
|
Dolor de cabeza |
12 |
1 |
0 |
|
Trastornos vasculares |
|||
|
Hipertensión |
10 |
5 |
0 |
|
a La reacción a la perfusión incluye los términos determinados por los investigadores para ser relacionados con la perfusión, ver debajo. b La neumonía también incluye los términos de neumonía estreptocócica y neumonía lobar. |
|||
|
Tabla 9: Anormalidades de laboratorio (≥10%) de Grado 3-4 emergentes del tratamiento |
|||
|
Daratumumab 16 mg/kg (N=156) |
|||
|
|
Todos los Grados (%) |
Grado 3 (%) |
Grado 4 (%) |
|
Anemia |
45 |
19 |
0 |
|
Trombocitopenia |
48 |
10 |
8 |
|
Neutropenia |
60 |
17 |
3 |
|
Linfopenia |
72 |
30 |
10 |
Reacciones a la perfusión: En los ensayos clínicos (tratamientos combinados y monoterapia; N=717) la incidencia de cualquier grado de reacción a la perfusión fue 46% con la primera perfusión de DARZALEX™, 2% con la segunda perfusión y 4% con perfusiones posteriores. Menos del 1% de los pacientes tuvo una reacción a la perfusión de Grado 3 con la segunda perfusión y posteriores perfusiones.
La mediana del tiempo de aparición de una reacción fue 1.5 horas (rango: 0.02 a 72.8 horas). La incidencia de la modificación de la perfusión debido a reacciones fue 41%. La media de las duraciones de la perfusión para la primera, segunda y posteriores infusiones fueron 7.0, 4.3 y 3.5 horas respectivamente.
Las reacciones a la perfusión severa (Grado 3) incluyeron broncoespasmo, disnea, edema laríngeo, edema pulmonar, hipoxia e hipertensión. Otras reacciones adversas a la perfusión (cualquier Grado, ≥5%) fueron congestión nasal, tos, escalofríos, irritación de garganta y vómitos.
Reactivación del virus del Herpes Zoster: La profilaxis para la reactivación del virus herpes zoster fue recomendada para pacientes en algunos ensayos clínicos de DARZALEX™. En estudios de monoterapia, el herpes zoster fue reportado en el 3% de los pacientes. En estudios aleatorizados de terapia combinada controlada, se reportó herpes zoster en el 2% de cada uno de los grupos de DRd y Rd respectivamente (Estudio 3) y en el 5% frente a 3% en los grupos de DVd y Vd respectivamente (Estudio 4).
Infecciones: En pacientes que reciben terapia combinada de DARZALEX™, las infecciones de Grado 3 ó 4 fueron reportadas con combinaciones de DARZALEX™ y terapias de base (DVd: 21%, Vd: 19%; DRd: 28%, Rd: 23%). La neumonía fue la infección severa más comúnmente reportada (Grado 3 ó 4) en los estudios. Las discontinuaciones del tratamiento fueron reportadas en el 3% frente a 2% de los pacientes en los grupos de DRd y Rd respectivamente y en el 4% frente 3% de los pacientes en los grupos de DVd y Vd respectivamente. Las infecciones fatales fueron reportadas en el 0.8% al 2% de los pacientes a través de los estudios, principalmente debido a neumonía y septicemia.
Inmunogenicidad: Como con todas las proteínas terapéuticas, existe el potencial de la inmunogenicidad. En ensayos clínicos de pacientes con mieloma múltiple tratados con DARZALEX™ como monoterapia o como terapias combinadas, ninguno de los 111 pacientes evaluables con monoterapia, y 1 (0.4%) de los 234 pacientes con terapias combinadas dieron positivo para anticuerpos anti-daratumumab. Este paciente recibió DARZALEX™ como terapia combinada, desarrollo anticuerpos neutralizantes transitorios contra daratumumab. Sin embargo, este ensayo tiene limitaciones para detectar anticuerpos anti-daratumumab en presencia de altas concentraciones de daratumumab; por lo tanto, la incidencia del desarrollo de anticuerpos podría no haber sido determinado de manera confiable.
Los datos de inmunogenicidad son altamente dependientes de la sensibilidad y especificidad de los métodos utilizados. Adicionalmente, la incidencia observada de un resultado positivo en un método de prueba puede estar influenciada por varios factores, incluyendo el manejo de muestras, tiempo de colección de la muestra, interferencia del fármaco, medicamento concomitante y enfermedad subyacente. Por lo tanto, la comparación de la incidencia de anticuerpos para daratumumab con la incidencia de anticuerpos a otros productos puede ser errónea.
Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de Farmacovigilancia: farmacovigilancia@digemid.minsa.gob.pe.
Interacciones con fármacos:
Efectos de daratumumab en pruebas de laboratorio:
Interferencia con pruebas de antiglobulina indirecta (prueba de Coombs Indirecta): Daratumumab se une al CD38 en los eritrocitos e interfiere con las pruebas de compatibilidad, incluyendo análisis de anticuerpos y concordancia cruzada. Los métodos de mitigación de la interferencia de daratumumab incluyen reactivo de tratamiento de los eritrocitos con ditiotreitol (DTT) para interrumpir el enlace daratumumab o genotipado. Dado que el sistema de grupos sanguíneos de Kell también es sensible al tratamiento con DTT, las unidades K-negativas deben suministrarse después de descartar o identificar los aloanticuerpos usando eritrocitos tratados con DTT.
Si se requiere de una transfusión de emergencia, los eritrocitos compatibles ABO/Rh sin realización de pruebas cruzadas pueden administrarse según las prácticas locales del banco de sangre.
Interferencia con las pruebas de electroforesis e inmunofijación de proteína sérica: Daratumumab puede ser detectada con las pruebas de electroforesis e inmunofijación de proteína sérica usada para el monitoreo de las inmunoglobulinas monoclonales de la enfermedad (proteína M). Esto puede conllevar a resultados falso positivo de la prueba electroforesis e inmunofijación de proteína sérica para pacientes con mieloma de proteína IgG kappa afectando la evaluación inicial de las respuestas completas de acuerdo al criterio del Grupo de Trabajo Internacional para Mieloma (IMWG). En pacientes con respuesta parcial persistente muy buena, considerar otros métodos para evaluar la profundidad de la respuesta.
ESTUDIOS CLÍNICOS:
Tratamiento combinado con lenalidomida y dexametasona: El Estudio 3, un ensayo aleatorizado y abierto, controlado con placebo en Fase 3, comparó el tratamiento de 16 mg/kg de DARZALEX™ en combinación con lenalidomida y dosis bajas de dexametasona (DRd) para el tratamiento con lenalidomida y dosis bajas de dexametasona (Rd) en pacientes con mieloma múltiple que habían recibido por lo menos una terapia previa. Lenalidomida (25 mg una vez al día oralmente en los días 1-21 de ciclos repetidos de 28 días (4 semanas) fue administrada con dosis bajas por vía oral o intravenosa de 40 mg/semana de dexametasona (o una dosis reducida de 20 mg/semana para pacientes > 75 años o índice de masa corporal (IMC) < 18.5). En los días de la perfusión con DARZALEX™, 20 mg de la dosis de dexametasona fue administrada como una medicación previa a la perfusión y el resto se administró el día después de la perfusión. Para pacientes con una dosis reducida de dexametasona, la dosis completa de 20 mg fue administrada como una medicación previa a la perfusión de DARZALEX™. Los ajustes de la dosis para lenalidomida y dexametasona fueron aplicados según la información de prescripción del fabricante. El tratamiento se continuó en ambos grupos hasta la progresión de la enfermedad o toxicidad inaceptable.
Un total de 569 pacientes fueron asignados al azar; 286 al grupo de DRd y 283 al grupo de Rd. Las características basales y de la enfermedad fueron similares entre DARZALEX™ y el grupo control. La mediana de la edad del paciente fue 65 años (rango de 34 a 89 años), el 11% fueron ≥ 75 años, 59% fueron varones; 69% Caucásicos, 18% Asiáticos y 3% Africanos Americanos. Los pacientes habían recibido una mediana de 1 línea previa de terapia. El sesenta y tres por ciento (63%) de los pacientes habían recibido trasplante autólogo previo de células progenitores. La mayoría de los pacientes (86%) recibió un inhibidor de proteasa previo, el 55% de los pacientes había recibido un agente inmunomodulador previo, incluyendo 18% de pacientes que habían recibido lenalidomida previa; y 44% de los pacientes habían recibido un inhibidor de proteasa previo y un agente inmunomodulador. En la basal, el 27% de los pacientes fueron refractarios a la última línea de tratamiento. Dieciocho por ciento (18%) de los pacientes fueron refractarios sólo al inhibidor de proteasa, y el 21% fueron refractarios a bortezomib. La eficacia fue evaluada por supervivencia libre de progresión basada en el criterio del Grupo de trabajo internacional para mieloma (IMWG).
El Estudio 3 demostró una mejora en la supervivencia libre de progresión en el grupo de DRd en comparación con el grupo de Rd; la mediana de la supervivencia libre de progresión no había sido alcanzada en el grupo DRd y fue 18.4 meses en el grupo de Rd (cociente de riesgo (HR)= 0.37; IC al 95%: 0.27, 0.52; p <0.0001), representando una reducción de 63% en el riesgo de la progresión de la enfermedad o muerte en pacientes tratados con DRd.
Figura 1: Curva Kaplan-Meter de supervivencia libre de progresión en el Estudio 3
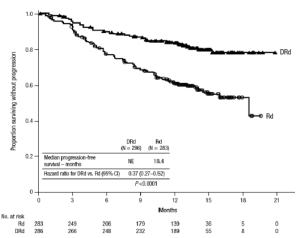
Los resultados de eficacia adicionales del Estudio 3 son presentados en la Tabla 10.
|
Tabla 10: Resultados adicionales de la eficacia del estudio 3a |
||
|
DRd (n=286) |
Rd (n=283) |
|
|
Respuesta general (sCR+CR+VGPR+PR) |
261 (91.3%) |
211 (74.6%) |
|
Valor-pb |
<0.0001 |
|
|
Respuesta completa estricta (sCR) |
51 (17.8%) |
20 (7.1%) |
|
Respuesta completa (CR) |
70 (24.5%) |
33 (11.7%) |
|
Respuesta parcial muy buen (VGRP) |
92 (32.2%) |
69 (24.4%) |
|
Respuesta parcial (PR) |
48 (16.8%) |
89 (31.4%) |
|
DRd = daratumumab – lenalidomida-dexametasona; Rd = lenalidomida-dexametasona; IC = intervalo de confianza. a Basado en intento de tratamiento a población b Valor-p de la Prueba del chi cuadrado de Cochran Mantel Haenszel |
||
En los respondedores, la mediana del tiempo para la respuesta fue 1 mes (rango: 0.9 a 13 meses) en el grupo de DRd y 1.1 meses (rango: 0.9 a 10 meses) en el grupo de Rd. La mediana de la duración de la respuesta no había sido alcanzada en el grupo de DRd (rango: 1+ a 19.8 + meses) y fue 17.4 meses (rango: 1.4 a 18.5+ meses) en el grupo de Rd.
Con un seguimiento mediano de 13.5 meses, se observaron 75 muertes; 30 en el grupo de DRd y 45 en el grupo Rd.
Tratamiento combinado con bortezomib y dexametasona: El estudio 4, un ensayo abierto, aleatorizado, controlado con activo, de Fase 3 comparó el tratamiento de 16 mg/kg de DARZALEX™ en combinación con bortezomib y dexametasona (DVd), para el tratamiento con bortezomib y dexametasona (Vd). Bortezomib fue administrado por inyección SC o perfusión IV a una dosis de 1.3 mg/m2 del área de la superficie corporal dos veces por semana por dos semanas (Días 1, 4, 8 y 11) de ciclos de tratamiento repetidos de 21 días (3 semanas) por un total de 8 ciclos. Dexametasona fue administrada oralmente a una dosis de 20 mg los días 1, 2, 4, 5, 8, 9, 11 y 12 de cada uno de los 8 ciclos de bortezomib (80 mg/semana por dos de tres semanas del ciclo de bortezomib) o a una dosis reducida de 20 mg/semana para pacientes > 75 años, con IMC < 18.5, diabetes mellitus mal controlada o intolerancia previa a la terapia de esteroides. En los días de la perfusión de DARZALEX™, 20 mg de la dosis de dexametasona fue administrada como una medicación previa a la perfusión. Para pacientes con una dosis reducida de dexametasona, la dosis completa de 20 mg fue administrada como una medicación previa a la perfusión de DARZALEX™. Bortezomib y dexametasona fueron administradas por ciclos 8 ciclos de 3 semanas en ambos grupos del tratamiento; mientras que DARZALEX™ fue administrado hasta la progresión de la enfermedad. Sin embargo, la dosis de 20 mg de dexametasona fue continuada como una medicación previa a la perfusión de DARZALEX™ en el grupo de DVd. Los ajustes de la dosis para bortezomib y dexametasona fueron aplicados según la información de prescripción del fabricante.
Un total de 498 pacientes fueron asignados al azar; 251 al grupo de DVd y 247 al grupo de Vd. Las características demográficas basales y de la enfermedad fueron similares entre DARZALEX™ y el grupo control. La edad mediana del paciente fue 64 años (rango 30 a 88 años); el 12% fueron ≥ 75 años, 57% fueron varones; 87% Caucásicos, 5% Asiáticos y 4% Africanos Americanos. Los pacientes habían recibido una mediana de 2 líneas de terapia previas y el 61% de los pacientes habían recibido trasplante autólogo previo de células progenitores. Sesenta y nueve por ciento (69%) de los pacientes habían recibido un inhibidor de proteasa previo (66% recibieron bortezomib) y 76% de los pacientes recibieron un agente inmunomodulador (42% recibieron lenalidomida). En el basal, el 32% de los pacientes fueron refractarios a la última línea de tratamiento y las proporciones de pacientes refractarios a cualquier terapia específica previa fueron en general bien equilibradas entre los grupos de tratamiento. Treinta y tres por ciento (33%) de los pacientes fueron refractarios solamente a un agente inmunomodulador, con 24% de los pacientes en el grupo de DVd y 33% de los pacientes en el grupo de Vd respectivamente refractarios a lenalidomida. La eficacia fue evaluada por supervivencia libre progresión basada en el criterio del Grupo de Trabajo Internacional para Mieloma.
El Estudio 4 demostró una mejoría en la supervivencia libre de progresión en el grupo de DVd en comparación con el grupo de Vd; la mediana de la supervivencia libre de progresión no se había alcanzado en el grupo de DVd y fue 7.2 meses en el grupo de Vd
(Cociente de riesgo [IC al 95%]: 0.39 (0.28, 0.53); valor-p <0.0001), representando una reducción del 61% en el riesgo de la progresión de la enfermedad o muerte para pacientes tratados con DVd frente a Vd.
Figura 2: Curva Kaplan-Meter de supervivencia libre de progresión en el Estudio 4
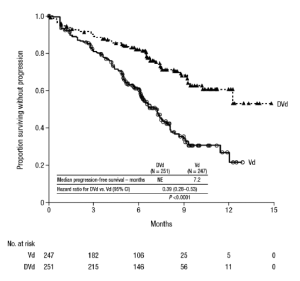
Los resultados adicionales de la eficacia del Estudio 4 son presentados en la Tabla 11.
|
Tabla 11: Resultados adicionales de la eficacia del Estudio 4 |
||
|
DVd (n=251) |
DVd (n= 247) |
|
|
Respuesta general (sCR+CR+VGPR+PR) |
199 (79.3%) |
148 (59.9%) |
|
Valor-pb |
<0.0001 |
|
|
Respuesta completa estricta (sCR) |
11 (4.4%) |
5 (2.0%) |
|
Respuesta completa (CR) |
35 (13.9%) |
16 (6.5%) |
|
Respuesta parcial muy buena (VGPR) |
96 (38.2%) |
47 (19.0%) |
|
Respuesta parcial (PR) |
57 (22.7%) |
80 32.4%) |
|
DVd = daratumumab – bortezomib – dexametasona; Vd = bortezomib – dexametasona; IC = Intervalo de confianza. a Basada en intento de tratamiento a población b Valor-p de la Prueba del chi cuadrado de Cochran Mantel Haenszel |
||
En los respondedores, el tiempo mediano a la respuesta fue 0.8 meses (rango: 0.7 a 4 meses) en el grupo de DVd y 1.5 meses (rango: 0.7 a 5 meses) en el grupo de Vd. La mediana de la duración de la respuesta no había sido alcanzada en el grupo de DVd (rango: 1.4+ a 14.1+ meses) y fue 7.9 meses (1.4+ a 12+ meses) en el grupo de Vd.
Con una mediana del seguimiento de 7.4 meses, se observaron 65 muertes; 29 en el grupo de DVd y 36 en el grupo de Vd.
Monoterapia: El Estudio 1, fue un ensayo abierto que evalúa la monoterapia de DARZALEX™ en pacientes con mieloma múltiple en recaída o refractario que habían recibido por lo menos 3 líneas de terapia previas incluyendo un inhibidor de proteasoma y un agente inmunomodulador o que eran doble refractarios a un inhibidor de proteasoma y a un agente inmunomodulador. En 106 pacientes, 16 mg/kg de DARZALEX™ fue administrado con medicación previa y posterior a la perfusión. El tratamiento continuó hasta la toxicidad inaceptable o progresión de la enfermedad.
La edad mediana del paciente fue 63.5 años (rango: 31 a 84 años), el 49% fueron varones y 79% fueron Caucásicos. Los pacientes habían recibido una mediana de 5 líneas de terapia previas. Ochenta por ciento de los pacientes habían recibido trasplante autólogo previo de células progenitores. Las terapias previas incluyeron bortezomib (99%), lenalidomida (99%), pomalidomida (63%) y carfilzomib (50%). En el basal, el 97% de los pacientes fueron refractarios a la última línea del tratamiento, 95% fueron refractarios a ambos, un inhibidor de proteasoma (IP) y agente inmunomodulador, y 77% fueron refractarios a agentes alquilantes.
Los resultados de la eficacia se basaron sobre la velocidad de respuesta general por la evaluación del Comité de Revisión Independiente utilizando los criterios del IMWG (ver Tabla 12).
|
Tabla 12: Resultados de la eficacia para el Estudio 1 |
|
|
N= 106 |
|
|
Velocidad de respuesta general (ORR) IC al 95% (%) |
31 (29.2%) (20.8, 38.9) |
|
Respuesta completa estricta (sCR) |
3 (2.8%) |
|
Respuesta completa (CR) |
0 |
|
Respuesta parcial muy buena (MBRP) |
10 (9.4%) |
|
Respuesta parcial (PR) |
18 (17.0%) |
|
ORR = sCR +CR+VGPR+PR IC = intervalo de confianza |
|
La mediana del tiempo a la respuesta fue 1 mes (rango: 0.9 a 5.6 meses). La mediana de la duración de la respuesta fue 7.4 meses (rango: 1.2 a 13.1 + meses).
El Estudio 2 fue un ensayo abierto de escalación de dosis evaluando la monoterapia de DARZALEX™ en pacientes con mieloma múltiple en recaída o refractario que habían recibido por lo menos 2 terapias citorreductivas diferentes. En 42 pacientes, 16 mg/kg de DARZALEX™ fue administrado con medicación previa y posterior a la perfusión. El tratamiento continuó hasta toxicidad inaceptable o progresión de la enfermedad.
La mediana de la edad del paciente fue 64 años (rango: 44 a 76 años), el 64% fueron varones y 76% fueron Caucásicos. Los pacientes en el estudio habían recibido una mediana de 4 líneas de terapia previa. Setenta y cuatro por ciento de los pacientes habían recibido trasplante autólogo previo de células progenitores. Las terapias previas incluyeron bortezomib (100%), lenalidomida (95%), pomalidomida (36%) y carfilzomib (19%). En la basal, el 76% de los pacientes fueron refractarios a la última línea del tratamiento, 64% de los pacientes fueron refractarios a ambos, un inhibidor de proteosoma y un agente inmunomodular, y 60% de los pacientes fueron refractarios a agentes alquilantes.
La velocidad de respuesta general fue 36% (IC al 95%: 21.6, 52.0%) con 1 CR y 3 VGPR. La mediana del tiempo a la respuesta fue 1 mes (rango: 0.5 a 3.2 meses). La mediana de la duración de la respuesta no fue estimable (rango: 2.2 a 13.1 + meses).
TOXICOLOGÍA NO CLÍNICA:
Carcinogénesis, mutagénesis, deterioro de la fertilidad: No se han realizado estudios de genotoxicidad o carcinogenicidad con daratumumab. No se han realizado estudios en animales para evaluar los efectos potenciales de daratumumab sobre la reproducción o desarrollo, o para determinar los efectos potenciales sobre la fertilidad en machos o hembras.
ADVERTENCIAS Y PRECAUCIONES:
Reacciones a la perfusión: DARZALEX™ puede causar reacciones severas a la perfusión. Aproximadamente la mitad de los pacientes experimentó una reacción, la mayoría durante la primera perfusión.
Las reacciones a la perfusión pueden ocurrir con perfusiones posteriores. Casi todas las reacciones ocurrieron durante la perfusión o dentro de las 4 horas de completar DARZALEX™. Antes de la introducción del medicamento de post-perfusión en ensayos clínicos, las reacciones a la perfusión ocurrieron hasta 48 horas después de la perfusión.
Han ocurrido reacciones graves, incluyendo broncoespasmo, hipoxia, disnea, hipertensión, edema laríngeo y edema pulmonar. Los signos y síntomas pueden incluir síntomas respiratorios, tales como congestión nasal, tos, irritación de la garganta, así como escalofríos, vómitos y náuseas. Los síntomas menos comunes fueron sibilancias, rinitis alérgica, pirexia, malestar en el pecho, prurito e hipotensión (ver sección Reacciones adversas).
Premedicar a los pacientes con antihistamínicos, antipiréticos y corticosteroides. Monitorear a los pacientes frecuentemente durante la toda la perfusión. Interrumpir la perfusión de DARZALEX™ por reacciones de cualquier severidad e instituir manejo médico según sea necesario. Discontinuar permanentemente la terapia de DARZALEX™ por reacciones que amenacen la vida (Grado 4). Para pacientes con reacciones de Grado 1, 2 ó 3, reducir la velocidad de la perfusión al reiniciar la perfusión (ver sección Dosis y administración).
Para reducir el riesgo de las reacciones retardadas a la perfusión, administrar corticosteroides orales a todos los pacientes tras las infusiones de DARZALEX™ (ver sección Dosis y administración). Los pacientes con un antecedente de enfermedad pulmonar obstructiva crónica pueden requerir medicamentos post-perfusión adicionales para manejar las complicaciones respiratorias. Considerar la prescripción de broncodilatadores de corta y larga acción y corticosteroides inhalados para pacientes con enfermedad pulmonar obstructiva crónica.
Interferencia con la prueba serológica: Daratumumab se une al CD38 en los eritrocitos y da como resultado una prueba antiglobulina indirecta positiva (prueba de Coombs indirecta). La prueba antiglobulina indirecta positiva mediada con daratumumab puede persistir hasta por 6 meses luego de la última perfusión de daratumumab. Daratumumab unido a los eritrocitos enmascara la detección de anticuerpos a antígenos menores en el suero del paciente. La determinación del tipo sanguíneo ABO y Rh del paciente no se ve afectada (ver sección Interacciones con fármacos).
Notificar a los centros de transfusión sanguínea de esta interferencia con las pruebas serológicas e informar a los bancos de sangre que un paciente ha recibido DARZALEX™. Tipificar y analizar a los pacientes antes de iniciar DARZALEX™.
Neutropenia: DARZALEX™ puede incrementar la neutropenia inducida por la terapia de base (ver sección Reacciones adversas).
Monitorear los recuentos completos de células sanguíneas periódicamente durante el tratamiento según la información de prescripción del fabricante para las terapias de base. Monitorear a los pacientes con neutropenia por signos de infección. La dosis de DARZALEX™ puede ser requerida para permitir la recuperación de los neutrófilos. No se recomienda ninguna reducción de la dosis de DARZALEX™. Considerar un cuidado de soporte con factores de crecimiento.
Trombocitopenia: DARZALEX™ puede incrementar la trombocitopenia inducida por la terapia de base (ver sección Reacciones adversas).
Monitorear los recuentos completos de células sanguíneas periódicamente durante el tratamiento según la información de prescripción del fabricante para las terapias de base. El retraso de la dosis de DARZALEX™ puede ser requerido para permitir la recuperación de las plaquetas. No se recomienda una reducción de la dosis de DARZALEX™. Considerar el cuidado de soporte con transfusiones.
Interferencia con la determinación de la respuesta completa: Daratumumab es un anticuerpo monoclonal humano IgG kappa que puede ser detectado en la electroforesis de proteínas séricas (EPS) y en los ensayos de inmunofijación (IFE) utilizados para el monitoreo clínico de la proteína M endógena. (Ver sección Interacciones con fármacos). Esta interferencia puede influir en la determinación de la respuesta completa y en la progresión de la enfermedad en algunos pacientes con mieloma de proteína IgG kappa.
Efectos sobre la capacidad para conducir y utilizar máquinas: La influencia de DARZALEX™ sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. Sin embargo, se ha notificado cansancio en pacientes que usan daratumumab y esto se debe tener en cuenta cuando se conduzca o se utilicen máquinas.
DOSIS Y ADMINISTRACIÓN:
Dosis recomendada y esquema:
• Administrar medicamentos de pre-perfusión y post-perfusión (ver sección Dosis y administración)
• Administrar únicamente como una perfusión intravenosa luego de la dilución en inyección de cloruro de sodio USP al 0.9% (ver sección Dosis y administración).
• DARZALEX™ debe ser administrado por un profesional de la salud, con acceso inmediato al equipo de emergencia y al soporte médico apropiado para manejar reacciones a la perfusión si éstas ocurren (ver sección Advertencias y precauciones).
Monoterapia y terapia combinada con lenalidomida y dosis bajas de dexametasona (régimen de ciclo de 4 semanas): La dosis recomendada de DARZALEX™ es 16 mg/kg de peso corporal actual administrado como una perfusión intravenosa según el siguiente esquema de dosis en la Tabla 1:
|
Tabla 1: Esquema de dosis de DARZALEX™ para monoterapia y en combinación con lenalidomida (régimen de dosis del ciclo de 4 semanas) |
|
|
Semanas |
Esquema |
|
Semanas 1 a 8 |
Semanalmente (total de 8 dosis) |
|
Semanas 9 a 24a |
Cada dos semanas (total de 8 dosis) |
|
Semana 25 en adelante hasta la progresión de la enfermedadb |
Cada cuatro semanas |
|
a Primera dosis del esquema de la dosis de cada 2 semanas es administrada en la semana 9 b Primera dosis del esquema de la dosis de cada 4 semanas es administrada en la semana 25. |
|
Para instrucciones de dosis de los agentes de combinación administrados con DARZALEX™, ver sección Estudios clínicos y la información de prescripción del fabricante.
Terapia combinada con bortezomib y dexametasona (régimen del ciclo de 3 semanas):
La dosis recomendada de DARZALEX™ es 16 mg/kg del peso corporal actual administradocomo una perfusión intravenosa según el siguiente esquema de dosis en la Tabla 2:
|
Tabla 2: Esquema de dosis de DARZALEX™ con bortezomib (régimen de dosis del ciclo de 3 semanas) |
|
|
Semanas |
Esquema |
|
Semanas 1 a 9 |
Semanalmente (total de 9 dosis) |
|
Semanas 10 a 24a |
Cada tres semanas (total de 5 dosis) |
|
Semana 25 en adelante hasta progresión de la enfermedad b |
Cada cuatro semanas |
|
a Primera dosis de cada esquema de dosis de cada 3 semanas es administrada en la semana 10 b Primera dosis de cada esquema de dosis de cada 4 semanas es administrada en la semana 25. |
|
Para instrucciones de dosis de agentes de combinación administrados con DARZALEX™ ver sección Estudios clínicos y la información de prescripción del fabricante.
Dosis de DARZALEX™ omitidas: Si se omite una dosis planificada de DARZALEX™, administrar la dosis lo antes posible y ajustar el esquema de la dosificación, manteniendo el intervalo del tratamiento.
Velocidad de perfusión y tratamiento de las reacciones a la perfusión: Administrar la perfusión de DARZALEX™ intravenosamente en la velocidad de perfusión descrita en la Tabla 3. Considerar la escalada de incrementos de la velocidad de la perfusión sólo en ausencia de reacciones a la perfusión.
|
Tabla 3: Velocidad s de perfusión para la administración de DARZALEX™ |
||||
|
Volumen de dilución |
Velocidad inicial (primera hora) |
Incremento de velocidada |
Velocidad máxima |
|
|
Primera perfusión |
1000 mL |
50 mL/hora |
50 mL/hora cada hora |
200 mL/hora |
|
Segunda perfusiónb |
500 mL |
50 mL/hora |
50 mL/hora cada hora |
200 mL/hora |
|
Infusiones subsiguientesc |
500 mL |
100 mL/hora |
50 mL/hora cada hora |
200 mL/hora |
|
a Considerar la escalada de incrementos de la velocidad de perfusión únicamente en ausencia de las reacciones a la perfusión. b Usar un volumen de dilución de 500 mL sólo si no hubiesen reacciones a la perfusión de Grado 1 (leve) o mayores durante las primeras 3 horas de la primera perfusión. De lo contrario, continuar utilizando un volumen de dilución de 1000 mL y las instrucciones para la primera perfusión. c Utilizar una velocidad inicial modificada para las perfusiones subsiguientes (esto es, tercera perfusión en adelante) sólo si no hubiese reacciones a la perfusión de Grado 1 (leve) o mayores durante una velocidad de perfusión final ≥ 100 mL/hora en las primeras dos infusiones. De lo contrario, continuar utilizando las instrucciones para la segunda perfusión. |
||||
Para las reacciones a la perfusión de cualquier grado/severidad, interrumpir inmediatamente la perfusión de DARZALEX™ y manejar los síntomas. El tratamiento de las reacciones a la perfusión puede requerir además la reducción de la velocidad de la perfusión, o la discontinuación del tratamiento con DARZALEX™ según se indica a continuación (ver sección Advertencias y Precauciones)
• Grado 1-2 (leve a moderado): Una vez que los síntomas de la reacción se resuelven, reanudar la perfusión a no más de la mitad de la velocidad a la que se produjo la reacción. Si el paciente no experimenta ningún síntoma de la reacción, la escalada de la velocidad de perfusión puede reanudarse en incrementos e intervalos según sea clínicamente apropiado hasta la velocidad máxima de 200 mL/hora (Tabla 3).
• Grado 3 (severo): Una vez que los síntomas de la reacción se resuelven, considerar reiniciar la perfusión a no más de la mitad de la velocidad a la que se produjo la reacción. Si el paciente no experimenta síntomas adicionales, reanudar la escalada de la velocidad de perfusión a incrementos e intervalos según se indican en la Tabla 3. Repetir el procedimiento anterior en caso de recurrencia de los síntomas de Grado 3. Discontinuar permanentemente DARZALEX™ a la tercera ocurrencia de una reacción a la perfusión de Grado 3 o mayor.
• Grado 4 (amenaza la vida): Discontinuar permanentemente el tratamiento con DARZALEX™.
Medicamentos concomitantes recomendados:
Medicamentos previos a la perfusión: Administrar los siguientes medicamentos de previo a la perfusión para reducir el riesgo de reacciones a la perfusión a todos los pacientes de 1 a 3 horas antes de cada perfusión de DARZALEX™.
• Corticosteroide (de acción prolongada o intermedia)
– Monoterapia: Metilprednisolona 100 mg, o equivalente, administrada intravenosamente. Después de la segunda perfusión, la dosis de corticosteroide puede ser reducida (60 mg de metilprednisolona intravenosa u oral).
– Terapia combinada: Administrar 20 mg de dexametasona antes de cada perfusión de DARZALEX™ (ver sección Estudios clínicos).
La dexametasona es administrada intravenosamente antes de la primera perfusión de DARZALEX™ y la administración oral puede ser considerada antes de las infusiones subsiguientes.
• Antipiréticos (650 a 1000 mg de acetaminofén oral)
• Antihistamínico (difenhidramina oral o intravenosa de 25 a 50 mg o equivalente).
Medicamentos posterior a la perfusión: Administrar medicación de post-perfusión para reducir el riesgo de reacciones a la perfusión retrasada a todos los pacientes según como sigue:
• Monoterapia: Administrar corticoesteroide oral (20 mg de metilprednisolona o dosis equivalente de un corticosteroide de acción prolongada o intermedia según las normas locales) en cada uno de los 2 días siguientes a todas las infusiones de DARZALEX™ (empezando el día después de la perfusión).
• Terapia combinada: Considerar la administración de bajas dosis de metilprednisolona oral (≤ 20 mg) o equivalente, el día después de la perfusión de DARZALEX™.
Sin embargo, si un corticosteroide específico de régimen de fondo (por ejemplo, dexametasona) es administrado el día después de la perfusión de DARZALEX™, los medicamentos de post-perfusión adicionales pueden no ser necesarios (ver sección Estudios Clínicos).
Además, para cualquier paciente con un antecedente de enfermedad pulmonar obstructiva crónica, considerar la prescripción de medicamentos de post-perfusión tales como broncodilatadores de corta y larga acción y corticosteroides inhalados. Después de la primera hora de infusiones, si el paciente no experimenta mayores reacciones a la perfusión, estos medicamentos adicionales de post-perfusión inhalados pueden ser discontinuados.
Profilaxis para la reactivación del herpes zoster: Iniciar la profilaxis antiviral para prevenir la reactivación del herpes zoster dentro de una semana luego de iniciar DARZALEX™ y continuar por 3 meses siguiendo el tratamiento (ver sección Reacciones adversas).
Modificaciones de la dosis: No se recomienda ninguna reducción de la dosis de DARZALEX™. Se puede requerir retraso de la dosis para permitir la recuperación del conteo de células sanguíneas en el caso de toxicidad hematológica (ver sección Advertencias y Precauciones). Para información relacionada a medicamentos administrados en combinación con DARZALEX™, ver la información de prescripción del fabricante.
Preparación para la administración: DARZALEX™ es para un solo uso.
Preparar la solución para la perfusión utilizando la técnica aséptica según como sigue:
• Calcular la dosis (mg), volumen total (mL) de la solución de DARZALEX™ requerida y el número de frascos de DARZALEX™ en base al peso corporal actual del paciente.
• Verificar que la solución de DARZALEX™ sea incolora a amarillo pálido. No utilizar si partículas opacas, decoloración u otras partículas extrañas están presentes.
• Eliminar un volumen de la inyección de cloruro de sodio USP al 0.9% de la bolsa/recipiente de la perfusión que es igual al volumen requerido de la solución DARZALEX™.
• Retirar la cantidad necesaria de solución de DARZALEX™ y diluir al volumen apropiado añadiéndolo a la bolsa/recipiente de perfusión que contiene inyección de cloruro de sodio USP al 0.9%, como se especifica en la Tabla 3 (ver sección Dosis y administración). Las bolsas/recipientes de la perfusión deben ser hechas ya sea de cloruro de polivinilo (PVC), polipropileno (PP), polietileno (PE) o mezcla de poliolefina (PP+PE). Diluir bajo condiciones asépticas apropiadas. Desechar cualquier porción no utilizada que haya quedado en el vial.
• Invierta suavemente la bolsa/recipiente para mezclar la solución. No agitar.
• Los productos farmacéuticos parenterales deben ser inspeccionados visualmente en busca de partículas y decoloración antes de la administración, siempre que la solución y el recipiente lo permitan. La solución diluida puede desarrollar partículas proteicas muy pequeñas, translúcidas a blancas, ya que daratumumab es una proteína. No utilizar si se observan partículas visiblemente opacas, decoloración o partículas extrañas.
• Debido a que DARZALEX™ no contiene un preservante, administrar la solución diluida inmediatamente a temperatura ambiente 15°C – 25 °C (59°F – 77°F) y en luz ambiente. La solución diluida puede conservarse a temperatura ambiente por un máximo de 15 horas (incluyendo el tiempo de perfusión).
• Si no se utiliza inmediatamente, la solución diluida puede ser almacenada antes de la administración por hasta 24 horas en condiciones refrigeradas de 2 °C – 8 °C (36 °F-46 °F) y protegida de la luz. No congelar.
Administración:
• Si es almacenada en el refrigerador, permitir que la solución llegue a temperatura ambiente. Administrar la solución diluida mediante perfusión intravenosa utilizando un equipo de perfusión equipado con un regulador de flujo y con un filtro en línea de polietersulfona (PES) de baja unión a la proteína, estéril, no pirogénico (tamaño de poro 0.22 ó 0.2 micrómetros). Los equipos de administración deben estar hechos de poliuretano (PU), polibutadieno (PBD), PVC, PP o PE.
• No almacenar ninguna porción no utilizada de la solución de la perfusión para la reutilización. Cualquier producto no utilizado o material de desecho deben eliminarse de acuerdo con los requisitos locales.
• No infundir DARZALEX™ concomitantemente en la misma línea intravenosa con otros agentes.
USO EN POBLACIONES ESPECÍFICAS:
Embarazo:
Resumen de riesgo: No existe información humana para informar un riesgo con el uso de DARZALEX™ durante el embarazo. No se han realizado estudios de animales. Sin embargo, existen consideraciones clínicas (ver sección Consideraciones clínicas). Se desconoce el riesgo de fondo estimado de los defectos del nacimiento y el aborto espontáneo para la población indicada. En la población general de USA, el riesgo de fondo estimado de los mayores defectos del nacimiento y aborto espontáneo en embarazos clínicamente reconocidos es 2-4% y 15-20%, respectivamente.
Consideraciones clínicas:
Reacciones adversas en el fetos/neonato: Los anticuerpos monoclonales de inmunoglobulina G1 (IgG1) son transferidos a través de la placenta. En base a su mecanismo de acción, DARZALEX™ puede causar mieloide fetal o depleción de células linfoides y disminución de la densidad ósea. Postergar la administración de las vacunas vivas a neonatos e infantes expuestos a DARZALEX™ en útero hasta que se complete una evaluación hematológica.
Datos:
Información animal: Los ratones que fueron genéticamente modificados para eliminar toda la expresión CD38 (ratones knockout CD38) habían reducido la densidad ósea en el nacimiento que se recuperó a los 5 meses de edad. En monos cynomolgus expuestos durante el embarazo a otros anticuerpos monoclonales que afectan a las poblaciones de leucocitos, los monos infantiles tuvieron una reducción reversible de los leucocitos.
Lactancia:
Resumen de riesgo: No existe información respecto a la presencia de daratumumab en la leche humana, los efectos sobre el infante amamantado o los efectos sobre la producción de leche. La IgG humana es conocida por estar presente en la lecha humana. La información publicada sugiere que los anticuerpos en la leche materna no ingresan a las circulaciones del neonato e infante en cantidades sustanciales.
Los beneficios para el desarrollo y la salud de la lactancia deben ser considerados junto con la necesidad clínica de la madre para DARZALEX™ y cualquier efecto adverso potencial de DARZALEX™ en el niño amamantado o de la condición materna subyacente.
Mujeres y varones con potencial reproductivo
Anticoncepción: A fin de evitar la exposición al feto, las mujeres con potencial reproductivo deben utilizar anticoncepción efectiva durante el tratamiento y por 3 meses luego del cese del tratamiento con DARZALEX™.
Uso pediátrico: No se ha establecido la seguridad y eficacia de DARZALEX™ en pacientes pediátricos.
Uso geriátrico: De los 156 pacientes que recibieron monoterapia de DARZALEX™ a la dosis recomendada, 45% tenían 65 años de edad o mayores, y el 10% tenían 75 años de edad o mayores. De los 561 pacientes que recibieron DARZALEX™ con varias terapias combinadas, 40% tenían 65 a 75 años de edad, y el 9% tenían 75 años de edad o mayores. No se observaron diferencias generales en la seguridad y eficacia entre estos pacientes y los pacientes más jóvenes (ver sección Estudios clínicos).
SOBREDOSIS:
Se desconoce la dosis de DARZALEX™ con la cual ocurre toxicidad severa.
En el caso de una sobredosis, monitorear a los pacientes por cualquier signo o síntoma de efectos adversos y proporcionar tratamiento de soporte apropiado.
INFORMACIÓN DE ORIENTACIÓN PARA EL PACIENTE:
Aconsejar al paciente que lea el inserto para el paciente aprobado por la Autoridad reguladora.
Reacciones de la perfusión: Aconsejar a los pacientes buscar atención médica inmediata por cualquiera de los siguientes signos y síntomas de las reacciones de la perfusión:
• Picazón, secreción o nariz tapada, escalofríos, náusea, irritación de garganta, tos, dolor de cabeza, dificultad para respirar (ver sección Advertencias y precauciones y Reacciones adversas).
Neutropenia: Aconsejar a los pacientes que si ellos tienen fiebre, ellos deben contactar a su profesional de la salud (ver sección Advertencias y precauciones y Reacciones adversas).
Trombocitopenia: Aconsejar a los pacientes informar a su profesional de la salud si ellos notan signos de moretones o sangrado (ver sección Advertencias y precauciones y Reacciones adversas).
Interferencia con pruebas de laboratorio: Aconsejar a los pacientes informar al profesional de la salud incluyendo los centros/personal de transfusión sanguínea que ellos están tomando DARZALEX™, en caso de una transfusión planeada (ver sección Advertencias y precauciones y Reacciones adversas).
Aconsejar a los pacientes que DARZALEX™ puede afectar los resultados de algunas pruebas utilizadas para determinar la respuesta completa en algunos pacientes y pruebas adicionales pueden ser necesarias para evaluar la respuesta (ver sección Advertencias y precauciones e Interacciones de fármacos).
DESCRIPCIÓN:
Daratumumab es un anticuerpo monoclonal humano de inmunoglobulina G1 kappa (IgGIk) contra el antígeno CD38, producido en una línea celular de mamífero (Ovario de Hamster Chino (OHC) utilizando tecnología de ADN recombinante. El peso molecular de daratumumab es aproximadamente 148 kDa.
DARZALEX™ es suministrado como una solución incolora a amarillo pálido libre de preservante para la perfusión intravenosa en viales de dosis única. El pH es 5.5. DARZALEX™ debe ser diluido con inyección de cloruro de sodio USP al 0.9% (ver sección Dosificación y administración).
FARMACOLOGÍA CLÍNICA:
Mecanismo de acción: CD38 es una glicoproteína de transmembrana (48 kDa) expresada sobre la superficie de las células hematopoyéticas, incluyendo mieloma múltiple y otros tipos de células y tejidos y tiene múltiples funciones, tales como adhesión mediada por el receptor, señalización y modulación de la actividad de la ciclasa e hidrolasa. Daratumumab es un anticuerpo monoclonal humano IgGlk (mAb) que se une a CD38 e inhibe el crecimiento de las células tumorales que expresan la CD38 mediante la inducción de la apoptosis directamente a través del entrecruzamiento mediado por el Fc así como por la lisis de células tumorales inmunitarias mediada por inmunidad a través de la citotoxicidad dependiente del complemento (CDC), citotoxicidad mediada de células dependientes de anticuerpos (ADCC) y fagocitosis celular dependiente de anticuerpos (ADCP). Un subgrupo de células supresoras derivadas de mieloide (CD38+MDSCs), células T reguladoras (CD38+Tregs) y células B (CD38+Bregs) son reducidas por daratumumab.
Farmacodinámica: Las células NK expresan CD38 y son susceptibles a la lisis celular mediada por daratumumab. Las disminuciones en los recuentos absolutos y los porcentajes de células NK totales (CD16+CD56+) y células NK activadas (CD16+CD56dim) en la sangre periférica entera y médula ósea fueron observados con el tratamiento de DARZALEX™.
Electrofisiología cardíaca: DARZALEX™ como proteína grande tiene una baja probabilidad de interacciones directas de los canales de iones. No hay evidencia de los datos no clínicos o clínicos que sugieran que DARZALEX™ tiene el potencial para retrasar la repolarización ventricular.
Farmacocinética: Sobre el rango de la dosis desde 1 a 24 mg/kg como monoterapia o 1 a 16 mg/kg de DARZALEX™ en combinación con otros tratamientos, los incrementos en el área bajo la curva tiempo-concentración (AUC) fueron más que a la proporcional a la dosis.
Tras la dosis recomendada de 16 mg/kg cuando DARZALEX™ fue administrada como monoterapia o en terapia combinada, el valor del promedio de la concentración máxima
sérica (Cmáx) al final de la dosis semanal, fue aproximadamente 2.7 a 3 veces superior en comparación al promedio de la Cmáx sérica después de la primera dosis. El promedio ± la desviación estándar a través de la concentración sérica (Cmin) al final de la dosis semanal fue 573 ± 332 ug/mL, cuando DARZALEX™ fue administrado como monoterapia y 502 ± 196 a 607 ± 231 ug/mL cuando DARZALEX™ fue administrado como terapia combinada. El estado estacionario de daratumumab se alcanzó aproximadamente 5 meses en el período de dosificación de cada 4 semanas (por la perfusión 21), y el promedio ± la desviación estándar de la proporción de la Cmáx en estado estacionario hasta la Cmáx luego de la primera dosis fue 1.6 +-0.5.
Distribución: A la dosis recomendada de 16 mg/kg, el promedio ± la desviación estándar del volumen de distribución central fue 4.7 ± 1.3 L cuando DARZALEX™ fue administrado como monoterapia y 4.4 ± 1.5 L cuando DARZALEX™ fue administrado como terapia combinada.
Eliminación: El aclaramiento de daratumumab disminuyó con el incremento de la dosis y con las dosis múltiples. Con la dosis recomendada de 16 mg/kg de DARZALEX™ como monoterapia, el promedio ± la desviación estándar del aclaramiento lineal se estimó en 171.4 ± 95.3 mL/día. El promedio ± la desviación estándar de la semivida terminal estimada asociado con el aclaramiento lineal fue 18 ± 9 días cuando DARZALEX™ fue administrada como monoterapia y 23 ± 12 días cuando DARZALEX™ fue administrada como terapia combinada.
Poblaciones específicas: Las siguientes características de la población no tienen efecto clínicamente significativo sobre la farmacocinética de daratumumab en pacientes administrados con DARZALEX™ como monoterapia o como terapia combinada: sexo, edad (31 a 84 años), leve (bilirrubina total 1 a 1.5 veces el límite superior de la normalidad (LSN) y cualquier alanina transaminasa (ALT) e insuficiencia hepática moderada (bilirrubina total 1.5 a 3 veces el LSN y cualquier ALT) o insuficiencia renal (aclaramiento de creatinina (CLcr) 15 - 89 mL/min). El efecto de la insuficiencia hepática severa (bilirrubina total > 3 veces el LSN y cualquier ALT) es desconocido. El aumento del peso corporal incrementó el volumen central de la distribución y el aclaramiento de daratumumab, soportando el régimen de dosificación basado en el peso corporal.
Interacciones de fármacos:
Efecto de otros fármacos sobre daratumumab: La coadministración de lenalidomida o bortezomib con DARZALEX™ no afectó la farmacocinética de daratumumab.
Efecto de daratumumab sobre otros fármacos: La coadministración de DARZALEX™ con bortezomib no afectó la farmacocinética de bortezomib.
NATURALEZA Y CONTENIDO DEL ENVASE:
Caja de cartón conteniendo 1 vial de vidrio incoloro tipo I de 5 mL con tapón de goma de color gris con sello de aluminio y tapa flip-off que contiene 100 mg de daratumumab.
Caja de cartón conteniendo 1 vial de vidrio tipo I incoloro de 20 mL con tapón de goma de color gris con sello de aluminio y tapa flip-off que contiene 400 mg de daratumumab


