

GILENYA
FINGOLIMOD
Cápsulas
Cápsulas , 0,5 Miligramos
DESCRIPCIÓN Y COMPOSICIÓN:
Forma farmacéutica: Cápsulas duras.
Sustancia farmacéutica:
Cada CÁPSULA contiene: 0,5 mg de fingolimod (como clorhidrato).
El clorhidrato de fingolimod es un análogo sintético de la esfingosina. Su nombre químico es clorhidrato de 2-amino-2-[2-(4-octilfenil)etil]propano-1,3-diol. Su fórmula molecular es C19H33NO2·HCl y tiene una masa molecular de 343,93.
El clorhidrato de fingolimod es un polvo cristalino, blanco o blanquecino, que se disuelve fácilmente en agua.
Principio activo: Fingolimod.
Excipientes: Manitol, estearato de magnesio, dióxido de titanio y gelatina.
INDICACIONES: GILENYA® está indicado como terapia modificadora del curso de la enfermedad para reducir la frecuencia de las recidivas y retrasar la progresión de la discapacidad en pacientes con esclerosis múltiple recidivante.
CONTRAINDICACIONES:
• Pacientes que en los últimos 6 meses experimentaron infarto de miocardio, angina inestable, accidente cerebrovascular, ataque isquémico transitorio, insuficiencia cardíaca descompensada que requiere hospitalización o insuficiencia cardíaca clase III/IV.
• Pacientes que se encuentren en tratamiento con medicamentos antiarrítmicos de clase Ia ó III no deben ser tratados con GILENYA® (ver Advertencias y Precauciones, Interacciones).
• Pacientes con presencia de bloqueo auriculoventricular (AV) de II grado o III grado de Mobitz tipo II o síndrome del seno enfermo, a menos que el paciente tenga un marcapaso en funcionamiento.
• Pacientes con intervalo QTc ≥ 500 mseg (ver Advertencias y Precauciones).
• Pacientes con disfunción hepática moderada o severa/cirrosis hepática (correspondiendo a clase B ó C de Child-Pugh) o infección de hepatitis B activa crónica o aguda no deben ser tratados con GILENYA®.
• Pacientes con edema macular.
• Niños y adolescentes no deben ser tratados con GILENYA®.
• GILENYA® es contraindicado durante el embarazo y lactancia.
EMBARAZO Y LACTANCIA:
Embarazo: GILENYA® está contraindicado durante el embarazo.
Los estudios en animales han revelado efectos tóxicos en la función reproductora, como pérdidas fetales o defectos orgánicos y, sobre todo, tronco arterial persistente y comunicación interventricular (véase Datos sobre toxicidad preclínica). Además, se sabe que el receptor afectado por el fingolimod (receptor de la esfingosina-1-fosfato) participa en la formación de vasos sanguíneos durante la embriogénesis. Aún no se sabe si se producirán malformaciones cardiovasculares en el ser humano. Se tienen muy pocos datos sobre el uso del fingolimod durante la gestación. En los ensayos clínicos, se registraron 20 embarazos en pacientes que tomaban fingolimod en el momento en que se les diagnosticó el embarazo, pero los datos son demasiado escasos como para sacar conclusiones sobre la inocuidad de GILENYA® durante la gestación.
Parto: No se dispone de datos sobre los efectos del fingolimod en el parto.
Mujeres en edad de procrear: Antes de iniciar el tratamiento con GILENYA®, se debe comunicar a las mujeres en edad de procrear los riesgos graves para el feto y la necesidad de adoptar medidas anticonceptivas eficaces durante el tratamiento con GILENYA®. Como el compuesto tarda unos 2 meses en eliminarse del organismo después de retirar el tratamiento (véase Advertencias y precauciones) el riesgo para el feto puede persistir y es necesario adoptar medidas anticonceptivas durante ese período.
Lactancia: El fingolimod pasa a la leche de los animales tratados durante la lactancia. Dada la posibilidad de que el fingolimod produzca reacciones adversas graves en el lactante, las mujeres que reciben GILENYA® no deben amamantar.
GILENYA® está contraindicado en mujeres lactantes.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MAQUINAS: La influencia de GILENYA® sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
REACCIONES ADVERSAS:
Resumen del perfil toxicológico: La población para el análisis de la seguridad de GILENYA® proviene de dos ensayos clínicos de fase III comparativos con placebo y de un ensayo clínico de fase III comparativo con tratamiento activo efectuado en pacientes con esclerosis múltiple remitente recidivante.
Incluye un total de 2431 pacientes que recibieron GILENYA® (en dosis de 0,5 o 1,25 mg). El estudio D2301 (FREEDOMS) fue un ensayo clínico comparativo con placebo, de 2 años de duración, efectuado en 854 pacientes con esclerosis múltiple que recibieron fingolimod (placebo: 418 pacientes). El estudio D2309 (FREEDOMS II) fue un ensayo clínico comparativo con placebo, de 2 años de duración, efectuado en 728 pacientes con esclerosis múltiple que recibieron fingolimod (placebo: 355 pacientes). En la base de datos conjunta de ambos estudios, las reacciones adversas más graves registradas con la dosis terapéutica recomendada de 0,5 mg fueron: infecciones, edema macular y bloqueos auriculoventriculares transitorios al inicio del tratamiento. Las reacciones adversas más frecuentes (registradas con una incidencia ≥10%) con la dosis de 0,5 mg fueron: Cefalea, elevación de enzimas hepáticas, diarrea, tos, gripe, sinusitis y dorsalgia. Los acontecimientos adversos registrados con una incidencia superior al 1% (con GILENYA® 0,5 mg) que llevaron a interrumpir el tratamiento eran elevaciones de ALT (2,2%).
En el estudio D2302 (TRANSFORMS), un ensayo clínico comparativo con interferón ß-1a, de 1 año de duración, en el que participaron 849 pacientes con esclerosis múltiple tratados con fingolimod, las reacciones adversas al fingolimod fueron generalmente similares a las de los estudios comparativos con placebo (teniendo en cuenta la diferente duración de los estudios).
Resumen tabulado de reacciones adversas: La Tabla 1 presenta la frecuencia de reacciones adversas observadas durante el análisis conjunto de los estudios comparativos con placebo FREEDOMS y FREEDOMS II.
Las reacciones adversas se enumeran con arreglo a la clase de órgano, aparato o sistema del MedDRA. Las categorías de frecuencia son las siguientes: muy frecuente (≥1/10); frecuente (≥ 1/100 a < 1/10); infrecuente (≥ 1/1000 a < 1/100).
Infecciones e infestaciones:
• Muy frecuentes: Infecciones virales por Influenza (11%), sinusitis (11%).
• Frecuentes: Bronquitis, herpes zoster, tiña versicolor.
• Poco frecuentes: Neumonía.
Trastornos de la sangre y del sistema linfático:
• Frecuentes: Leucopenia, linfopenia.
Trastornos del sistema nervioso:
• Muy frecuentes: Dolor de cabeza (25%).
• Frecuentes: Mareo, migraña.
• Raros: Síndrome de encefalopatía posterior reversible (SEPR)*
Trastornos oculares:
• Frecuentes: Visión borrosa.
• Poco frecuentes: Edema macular*
Trastornos cardiacos:
• Frecuentes: Bradicardia, bloqueo auriculoventricular.
Trastornos vasculares:
• Frecuentes: Hipertensión.
Trastornos respiratorios:
• Muy frecuentes: Tos (12%).
• Común: Disnea.
Trastornos gastrointestinales
• Muy frecuentes: Diarrea (13%).
Trastornos de la piel y del tejido subcutáneo:
• Frecuentes: Eczema, prurito.
Trastornos musculoesqueléticos:
• Muy frecuentes: Dolor de espalda (10%).
Trastornos generales:
• Frecuentes: Astenia.
Investigaciones:
• Muy frecuentes: Aumento de enzimas hepáticas (incrementos de ALT, GGT, AST) (15%).
• Frecuentes: Incremento de triglicéridos sanguíneos.
* No notificado en los ensayos FREEDOMS, FREEDOMS II y TRANSFORMS. La frecuencia se basa en una exposición estimada a fingolimod de 10000 pacientes en el total de ensayos.
Descripción de reacciones adversas seleccionadas:
Infecciones: En los ensayos clínicos de esclerosis múltiple, el porcentaje general de infecciones (65,1%) con la dosis de 0,5 mg fue semejante al del placebo. No obstante, la bronquitis, el herpes zóster y la neumonía resultaron más frecuentes en los pacientes tratados con GILENYA®. Se registraron infecciones graves en el 1,6% de los pacientes tratados con 0,5 mg de fingolimod y en el 1,4% de los pacientes del grupo del placebo.
Se han descrito muy ocasionalmente casos mortales de infección por el VZV con la coadministración prolongada de corticoesteroides (durante más de 5 días) para el tratamiento de las recidivas de la esclerosis múltiple, pero no se ha podido establecer una relación causal entre el tratamiento concomitante y el desenlace mortal. La coadministración de un tratamiento breve con corticoesteroides (de hasta 5 días de duración de acuerdo con los protocolos de los estudios) no aumentó la tasa general de infecciones en los pacientes tratados con fingolimod de los ensayos clínicos de fase III en comparación con el placebo (véanse Advertencias y precauciones e Interacciones).
Ha habido casos muy esporádicos de otras infecciones herpéticas con desenlaces mortales.
No obstante, no se ha confirmado su relación de causalidad con GILENYA®.
Edema macular: En los ensayos clínicos, se registraron casos de edema macular en el 0,5% de los pacientes tratados con la dosis recomendada de 0,5 mg de fingolimod (GILENYA®) y en el 1,1% de los pacientes tratados con la dosis más elevada de 1,25 mg.
En los ensayos clínicos de esclerosis múltiple, la mayoría de los casos ocurrieron en los primeros 3-4 meses de tratamiento. Algunos pacientes presentaban visión borrosa o menor agudeza visual, pero otros eran asintomáticos y se les hizo el diagnóstico durante una exploración oftalmológica ordinaria. El edema macular solía mejorar o desaparecía de forma espontánea después de suspender la administración del fármaco. No se ha evaluado el riesgo de recidiva tras una segunda exposición.
La incidencia de edema macular es mayor en los pacientes con esclerosis múltiple que tienen antecedentes de uveítis (es casi del 20% en los pacientes con antecedentes de uveítis y del 0,6% en los pacientes sin tales antecedentes).
Bradiarritmia: El inicio del tratamiento con GILENYA® produce una disminución transitoria de la frecuencia cardíaca y también puede asociarse a un retraso de la conducción auriculoventricular (véase Advertencias y precauciones).
En los ensayos clínicos de esclerosis múltiple, el descenso máximo medio de la frecuencia cardíaca se observó dentro de las 6 horas de la ingestión de la primera dosis, y con GILENYA® 0,5 mg se registraron disminuciones de la frecuencia cardíaca media, a juzgar por el pulso, de 8 latidos por minuto. La segunda dosis puede provocar una pequeña disminución adicional.
En raras ocasiones se observó una frecuencia cardíaca inferior a 40 latidos por minuto en pacientes tratados con GILENYA® 0,5 mg. La frecuencia cardíaca regresa a su valor inicial en el plazo de 1 mes de administración crónica.
En el programa clínico de esclerosis múltiple, se detectó un bloqueo auriculoventricular de primer grado (intervalo PR prolongado en el electrocardiograma) posterior al inicio del tratamiento en el 4,7% de los pacientes del grupo de GILENYA® 0,5 mg, en el 2,8% de los pacientes del grupo de interferón ß-1a intramuscular y en el 1,6% de los pacientes del grupo del placebo. También se detectó un bloqueo auriculoventricular de segundo grado en menos del 0,2% de los pacientes del grupo de GILENYA® 0,5 mg.
Desde la comercialización de GILENYA®, hubo comunicaciones aisladas de bloqueo auriculoventricular completo, de carácter transitorio y resolución espontánea, durante el período de observación de seis horas posterior a la administración de la primera dosis en pacientes tratados con GILENYA®. Los pacientes se recuperaban espontáneamente.
Los trastornos de la conducción observados tanto en los ensayos clínicos como durante la comercialización de GILENYA® suelen ser transitorios, asintomáticos y se resuelven en un plazo de 24 horas durante el tratamiento. Aunque la mayoría de los pacientes no necesitó intervención médica, en los ensayos clínicos, uno de los pacientes del grupo de 0,5 mg recibió isoprenalina contra un bloqueo auriculoventricular asintomático de segundo grado (Mobitz de tipo I).
Durante la comercialización de GILENYA®, en las 24 horas posteriores a la administración de la primera dosis, se han descrito sucesos aislados de inicio tardío tales como asístoles transitorias y muertes idiopáticas. La comedicación o las enfermedades prexistentes funcionaban como factores de confusión en tales casos. La relación de tales acontecimientos con GILENYA® es dudosa.
Presión sanguínea: En los ensayos clínicos de esclerosis múltiple, la dosis de 0,5 mg de fingolimod (GILENYA®) se asoció a un aumento leve de casi 1 mmHg (en promedio) de la tensión arterial media, que se manifestó al cabo de dos meses de tratamiento aproximadamente. Dicho aumento persistió con el tratamiento continuo. Se registró hipertensión en el 6,5% de los pacientes del grupo de
GILENYA® 0,5 mg y en el 3,3% de los pacientes que recibieron el placebo.
Función hepática: En pacientes con esclerosis múltiple tratados con GILENYA® se han registrado cifras elevadas de enzimas hepáticas, principalmente de alanina-transaminasa (ALT). En los ensayos clínicos, el 8,0% y el 1,8% de los pacientes tratados con GILENYA® 0,5 mg experimentaron un aumento asintomático de las concentraciones séricas de ALT igual o superior al triple del límite superior del intervalo normal de valores (≥3 x LSN) e igual o superior al quíntuple de dicho límite (≥5 x LSN), respectivamente, en comparación con las cifras de 1,9% y 0,9% obtenidas en el grupo del placebo, respectivamente. La mayoría de las elevaciones se produjeron en el curso de 6-9 meses. Las cifras de ALT se normalizaron en los dos meses posteriores a la retirada de GILENYA® aproximadamente. En los pocos pacientes que tuvieron elevaciones de ALT ≥5 x LSN y continuaron recibiendo GILENYA®, las cifras de ALT se normalizaron en aproximadamente 5 meses (véase Advertencias y precauciones).
Sistema respiratorio: Durante el tratamiento con fingolimod, se apreciaron disminuciones dosis dependientes leves del FEV1 (volumen espiratorio máximo en el primer segundo) y de la DLCO (capacidad de difusión pulmonar de monóxido de carbono) (ver Advertencias y Precauciones y Farmacocinética).
Episodios vasculares: En los ensayos clínicos de fase III, se han descrito casos esporádicos de enfermedad oclusiva de las arterias periféricas en pacientes que recibieron dosis elevadas de GILENYA® (1,25 o 5,0 mg). También se han comunicado casos esporádicos de ictus hemorrágicos e isquémicos con la dosis de 0,5 mg en los ensayos clínicos y durante la comercialización de GILENYA®, pero aún no se ha confirmado la relación de causalidad.
Neoplasmas cutáneos: En el estudio 2, el melanoma maligno y cada carcinoma celular basal ocurrió en 3 pacientes usando diariamente 0,5 mg de fingolimod (grupo interferón: Un carcinoma celular basal y un carcinoma celular escamoso). En el estudio 1, cada cuatro pacientes en el placebo y GILENYA® (0,5 mg diario) los grupos desarrollaron neoplasmas malignos cutáneos. Sin embargo no hubo acumulación clara de casos durante el tratamiento con GILENYA®, pacientes con riesgo de neoplasma maligno cutáneo deben someterse a evaluación dermatológica previo al inicio del tratamiento con GILENYA® y durante el curso del tratamiento.
Linfomas: Ha habido casos de linfoma en los ensayos clínicos y durante la comercialización de GILENYA®.
Los casos notificados eran de naturaleza heterogénea, pudiendo ser linfomas linfocíticos B o T. Su relación con GILENYA® sigue siendo dudosa.
INCOMPATIBILIDADES: No procede.
INTERACCIONES:
Interacciones farmacodinámicas: Los antineoplásicos, los inmunomoduladores o los inmunodepresores (como los corticoesteroides) no deben coadministrarse debido al riesgo de efectos aditivos en el sistema inmunitario (Ver Advertencias y precauciones). Las decisiones específicas sobre la posología y la duración del tratamiento simultáneo con corticoesteroides deben basarse en el criterio clínico. La coadministración de un tratamiento breve con corticoesteroides (de hasta 5 días de duración de acuerdo con los protocolos de los estudios) no aumentó la tasa general de infecciones en los pacientes tratados con fingolimod de los ensayos clínicos de fase III en comparación con el placebo (véanse Advertencias y precauciones e Reacciones adversas).
Asimismo, se debe tener precaución a la hora de sustituir un tratamiento a base de sustancias de acción prolongada que afectan el sistema inmunitario, como el natalizumab o la mitoxantrona, por GILENYA® (véase Advertencias y precauciones: Tratamiento previo con inmunodepresores o inmunomoduladores).
Cuando el fingolimod se usa con atenolol, se produce una reducción adicional de la frecuencia cardíaca igual al 15% al inicio del tratamiento con fingolimod, pero este efecto no se observa con el diltiazem. Debido a los posibles efectos aditivos sobre la frecuencia cardíaca, el tratamiento con GILENYA® no debe iniciarse en pacientes que reciben betabloqueantes, antagonistas del calcio capaces de disminuir la frecuencia cardíaca (como el verapamilo, el diltiazem o la ivabradina) u otras sustancias que puedan reducir dicha frecuencia (p.ej.: digoxina, inhibidores de la acetilcolinesterasa [AChEl], pilocarpina). Cuando se considere la posibilidad de tratamiento con GILENYA®, se debe pedir asesoramiento a un cardiólogo sobre la alternativa de usar medicamentos que no disminuyan la frecuencia cardíaca o una monitorización adecuada para iniciar el tratamiento (la cual debe durar toda la noche), véase Advertencias y precauciones y Dosificación/administración. GILENYA® es contraindicado en pacientes que toman medicamentos antiarrítmicos clase Ia o clase III. (Ver Contraindicaciones).
Durante el tratamiento con GILENYA® y hasta dos meses después del mismo, las vacunas pueden ser menos eficaces. El uso de vacunas atenuadas elaboradas con microbios vivos entraña un riesgo de infección y debe evitarse (véanse Reacciones adversas y Advertencias y precauciones).
Interacciones farmacocinéticas: El fingolimod es eliminado principalmente por medio del citocromo P450 4 F2 (CYP4F2) y posiblemente a través de otras isoformas del CYP4F. Los estudios in vitro en hepatocitos indicaron que el CYP3A4 puede contribuir al metabolismo del fingolimod en caso de inducción potente del CYP3A4.
Capacidad del fingolimod y del fosfato de fingolimod para inhibir el metabolismo de la comedicación: Los estudios de inhibición in vitro efectuados en microsomas hepáticos humanos y con sustratos analíticos específicos de enzimas metabólicas demuestran que el fingolimod y el fosfato de fingolimod tienen poca o ninguna capacidad para inhibir la actividad de las enzimas citocrómicas (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 o CYP4A9/11 [solo el fingolimod]). Por consiguiente, es improbable que el fingolimod y el fosfato de fingolimod reduzcan la depuración de los fármacos cuya eliminación metabólica se produce básicamente a través de las principales isoformas de las enzimas CYP.
Capacidad del fingolimod y del fosfato de fingolimod para inducir su propio metabolismo o el metabolismo de la comedicación: Se examinó la capacidad del fingolimod para inducir el ARNm del CYP3A4, CYP1A2, CYP4F2 o de la ABCB1 (glucoproteína P) y la actividad del CYP3A, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 o del CYP4F2 en hepatocitos primarios humanos. El fingolimod no indujo el ARNm ni la actividad de las diferentes enzimas citocrómicas o de la ABCB1 con respecto al vehículo de referencia. Por consiguiente, no cabe esperar una inducción clínicamente importante de las enzimas citocrómicas analizadas CYP450 o de la ABCB1 (glucoproteína PgP) por parte del fingolimod a las concentraciones terapéuticas. Los experimentos in vitro no aportaron indicios de inducción citocrómica por parte del fosfato de fingolimod.
Capacidad del fingolimod y del fosfato de fingolimod para inhibir el transporte activo de la comedicación: Los datos in vitro indican que no es probable que el fingolimod o el fosfato de fingolimod inhiban la captación de los cofármacos o productos biológicos que son transportados por los polipéptidos transportadores de aniones orgánicos (OATP1B1, OATP1B3) o el polipéptido cotransportador de taurocolato sódico (NTCP). Tampoco cabe esperar que, en concentraciones terapéuticas, inhiban la salida (al espacio extravascular) de los cofármacos o productos biológicos que son transportados por la proteína de resistencia al cáncer de mama (BCRP), la bomba de secreción de sales biliares (BSEP), la proteína 2 asociada a multirresistencia farmacológica (MRP2) o la glucoproteína P (P-gP).
Anticonceptivos orales: La administración diaria de 0,5 mg de fingolimod con anticonceptivos orales (etinilestradiol y levonorgestrel) no alteró la exposición a estos últimos. La exposición al fingolimod y al fosfato de fingolimod fue similar a la registrada en estudios previos. No se han realizado estudios de interacción con anticonceptivos orales que contienen otros progestágenos, pero no cabe esperar que el fingolimod altere su exposición.
Ciclosporina: La farmacocinética del fingolimod en dosis únicas permaneció invariable durante su administración con ciclosporina en el estado estacionario, y la farmacocinética de la ciclosporina en el estado estacionario no fue alterada por la administración de dosis únicas o repetidas de fingolimod (durante 28 días). Estos datos indican que es improbable que el fingolimod reduzca o aumente la depuración de fármacos que se eliminan principalmente por medio del CYP3A4, y que tampoco es probable que la inhibición del CYP3A4 reduzca la depuración del fingolimod. La inhibición potente de transportadores, como la glucoproteína P, la MRP2 o el OATP1BI, no afecta el destino del fingolimod.
Ketoconazol: La coadministración de 200 mg de ketoconazol dos veces al día en el estado estacionario y de una dosis única de 5 mg de fingolimod produjo un aumento moderado del AUC del fingolimod o del fosfato de fingolimod (cifras 1,7 veces mayores) debido a la inhibición del
CYP4F2.
Isoprenalina, atropina, atenolol y diltiazem: La administración conjunta de isoprenalina o atropina no alteró la exposición al fosfato de fingolimod o al fingolimod administrado en dosis únicas. Además, la farmacocinética tras dosis únicas del fingolimod y del fosfato de fingolimod y la farmacocinética en el estado estacionario del atenolol y el diltiazem permanecieron invariables cuando los dos últimos fármacos se administraron con fingolimod.
Carbamazepina: La coadministración de 600 mg de carbamazepina dos veces al día en el estado estacionario y de una dosis única de 2 mg de fingolimod ejerció un efecto débil sobre los AUC del fingolimod o del fosfato de fingolimod, disminuyendo ambos en un 40%, indicando que el uso concomitante de carbamazepina puede reducir la eficacia de fingolimod.
Otro inductor potente enzimático CYP3A4 (por ejemplo rifampicina, fenobarbital, fenitoína, oxcarbazepina, efavirenz y la hierba de San Juan) puede reducir el AUC de fingolimod y sus metabolitos al menos con una extensión comparable y puede comprometer la eficacia de fingolimod si es tomado conjuntamente.
Análisis farmacocinético poblacional de posibles interacciones farmacológicas: Un análisis farmacocinético poblacional realizado en pacientes con esclerosis múltiple no arrojó indicios de que la fluoxetina y la paroxetina (inhibidores potentes del CYP2D6) afecten de forma significativa las concentraciones de fingolimod o de fosfato de fingolimod. La administración de carbamazepina reduce los niveles de fosfato de fingolimod por menos al 30%. Además, las siguientes sustancias de prescripción común no afectan de forma clínicamente importante (≤20%) la concentración de fingolimod o de fosfato de fingolimod: Baclofeno, gabapentina, oxibutinina, amantadina, modafinilo, amitriptilina, pregabalina, corticoesteroides y anticonceptivos orales.
Pruebas de laboratorio: El fingolimod disminuye la cantidad de linfocitos sanguíneos al propiciar su redistribución entre órganos linfoides secundarios, de modo que no es posible utilizar la cifra de linfocitos en sangre periférica para evaluar el estado de los subgrupos de linfocitos de un paciente tratado con GILENYA®.
Las pruebas de laboratorio que exigen el uso de células mononucleadas circulantes (linfocitos) requieren un volumen mayor de sangre a causa del menor número de linfocitos circulantes.
ESTUDIOS CLÍNICOS
La eficacia de GILENYA® fue comprobada en dos estudios de pacientes con esclerosis múltiple remitente recidivante que recibieron 0,5 y 1,25 mg de fingolimod (GILENYA®) una vez al día. Ambos estudios incluyeron pacientes que habían experimentado al menos dos recidivas clínicas en los dos años anteriores a la aleatorización, o al menos una recidiva clínica en el año precedente a dicha aleatorización, y que presentaban puntuaciones de entre 0 y 5,5 en la Escala Ampliada del Estado de Discapacidad (EDSS). Tras el registro de GILENYA® se llevó a cabo un tercer estudio en la misma población de pacientes.
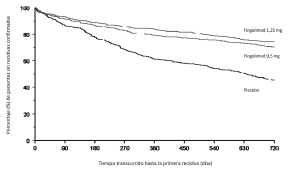
Estudio D2301 (FREEDOMS): El estudio D2301 (FREEDOMS) fue un ensayo clínico de fase III comparativo con placebo, aleatorizado, de doble enmascaramiento y de dos años de duración, efectuado en pacientes con esclerosis múltiple remitente recidivante que no habían recibido interferón ß ni acetato de glatirámero por lo menos durante 3 meses antes del estudio, ni natalizumab por lo menos durante 6 meses antes del estudio. La edad mediana fue de 37 años, la duración mediana de la enfermedad, de 6,7 años, y la puntuación mediana de la EDSS al inicio, de 2,0. Se aleatorizó a los pacientes de forma que recibiesen GILENYA® 0,5 mg (n=425), GILENYA® 1,25 mg (n=429) o el placebo (n=418) durante 24 meses. La duración mediana de tratamiento fue de 717 días (GILENYA® 0,5 mg), 715 días (GILENYA® 1,25 mg) y 718,5 días (placebo).
El criterio principal de valoración fue la tasa anual de recaídas.
La tasa anualizada de recidivas (TAR) fue significativamente menor en los pacientes del grupo de GILENYA® que en los del placebo. El criterio de valoración secundario clave fue el tiempo transcurrido hasta la progresión de la discapacidad confirmada durante 3 meses, que se determinó a través de un incremento de al menos un punto en la EDSS con respecto al inicio (o de un incremento de 0,5 puntos en los pacientes con EDSS inicial de 5,5) de forma sostenida durante 3 meses. El tiempo transcurrido hasta la progresión de la discapacidad confirmada durante tres meses se alargó considerablemente con el tratamiento con GILENYA® en comparación con el placebo. No se apreciaron diferencias significativas entre las dosis de 0,5 y 1,25 mg en ningún criterio de valoración.
Los resultados de este estudio se presentan en la Tabla 2 y las Figuras 1.
|
Tabla 2. Resultados de la IRM y clínicos del estudio FREEDOMS |
||
|
GILENYA® 0,5 mg |
Placebo |
|
|
Criterios clínicos |
N=425 |
N=418 |
|
Tasa anualizada de recidivas (criterio principal) |
0,18 (p<0,001*) |
0,40 |
|
Reducción relativa (porcentaje) |
54 |
|
|
Porcentaje de pacientes sin recidivas al cabo de 24 meses |
70,4 (p<0,001*) |
45,6 |
|
Riesgo de progresión de la discapacidad |
||
|
Cociente de riesgos instantáneos (IC del 95%) (confirmado durante tres meses) |
0,70 (0,52, 0,96) (p=0,024*) |
|
|
Cociente de riesgos instantáneos (IC del 95%) (confirmado durante seis meses) |
0,63 (0,44, 0,90) (p=0,012*) |
|
|
Criterios basados en la IRM |
||
|
Número de lesiones T2 nuevas o expansivas |
n=370 |
n=339 |
|
Número mediano (medio) en 24 meses |
0,0 (2,5) (p<0,001*) |
5,0 (9,8) |
|
Número de lesiones realzadas con Gd |
n=369 (mes 24) |
n=332 (mes 24) |
|
Número mediano (medio) al |
||
|
Mes 6 |
0,0 (0,2) |
0,0 (1,3) |
|
Mes 12 |
0,0 (0,2) |
0,0 (1,1) |
|
Mes 24 |
0,0 (0,2) (p<0,001* a cada tiempo valorado) |
0,0 (1,1) |
|
Cambio porcentual del volumen total de lesiones en T2 |
n=368 |
n=339 |
|
Cambio porcentual mediano (medio) en 24 meses |
-1,7 (10,6) (p<0,001*) |
8,6 (33,8) |
|
Cambio del volumen de lesiones hipointensas en T1 |
n=346 |
n=305 |
|
Cambio porcentual mediano (medio) en 24 meses |
0,0 (8,8) (p<0,012*) |
1,6 (50,7) |
|
Cambio porcentual del volumen cerebral |
n=357 |
n=331 |
|
Cambio porcentual mediano (medio) en 24 meses |
-0,7 (-0,8) (p<0,001*) |
-1,0 (-1,3) |
Todos los análisis de los criterios clínicos fueron análisis por intención de tratar (análisis IDT). En los análisis de los criterios basados en la IRM se usó un archivo evaluable de datos.
* Indica significación estadística con respecto al placebo con un nivel de confianza bilateral de 0,05.
Determinación de los valores de p: Tasa anualizada de recidivas (TAR) general, por regresión binomial negativa, con ajuste por tratamiento, país, número de recidivas en los 2 años previos y EDSS inicial; porcentaje de pacientes sin recidivas, por el modelo de regresión logística, con ajuste por tratamiento, país, número de recidivas en los 2 años previos y EDSS inicial; tiempo transcurrido hasta la progresión de la discapacidad confirmada a lo largo de 3 o 6 meses, por el modelo de riesgos proporcionales de Cox, con ajuste por tratamiento, país, EDSS inicial y edad; lesiones T2 nuevas o expansivas, por regresión binomial negativa, con ajuste por tratamiento y país; lesiones realzadas con Gd, por análisis de covarianza (ANCOVA) ordinal, con ajuste por tratamiento, país y número inicial de lesiones realzadas con Gd; y cambio porcentual del volumen de lesiones y del volumen cerebral, por ANCOVA ordinal, con ajuste por tratamiento, país y el valor inicial correspondiente.
Figura 1. Gráfico de Kaplan-Meier del tiempo transcurrido hasta la primera recidiva confirmada, hasta el mes 24. Estudio FREEDOMS (población por IDT)
Los pacientes que completaron el estudio FREEDOMS (D2301) tenían la posibilidad de participar en la ampliación del mismo (D2301E1) con doble enmascaramiento. Así pues, 920 pacientes del estudio principal pasaron a la fase de ampliación y recibieron fingolimod (n=331 continuaron en el grupo de 0,5 mg, 289 siguieron en el grupo de 1,25 mg, 155 del grupo del placebo pasaron a recibir 0,5 mg de fingolimod y 145 del grupo del placebo pasaron a recibir 1,25 mg de fingolimod). En 811 de estos pacientes (el 88,2%), el seguimiento fue de por lo menos 18 meses durante la fase de ampliación.
Al mes 24 de la ampliación del estudio, los pacientes que en el estudio principal habían recibido el placebo presentaban reducciones de la TAR del 55% tras su pase al grupo de fingolimod 0,5 mg (cociente de TAR: 0,45, IC del 95%: 0,32 – 0,62, p<0,001). La TAR de los pacientes que en el estudio principal habían recibido 0,5 mg de fingolimod siguió siendo baja durante la ampliación del estudio (TAR de 0,10 en la ampliación del estudio).
Entre los meses 24 y 36, tasa anualizada de recidivas (TAR) fue 0,17 para pacientes con 0,5 mg de fingolimod en el estudio principal quien continuó para tomar 0,5 mg en el estudio de extensión (0.21 en el estudio principal). Pacientes que cambiaron de placebo a 0,5 mg de fingolimod tuvieron un TAR de 0.22 (0.42 en el estudio principal).
Estudio D2309 (FREEDOMS II): Se obtuvieron resultados comparables en una réplica de ensayo con esclerosis múltiple remitente recidivante de Fase III, randomizado, doble ciego, controlado con placebo en 1083 pacientes al estudio D2301. Este estudio fue completado después de la aprobación de fingolimod.
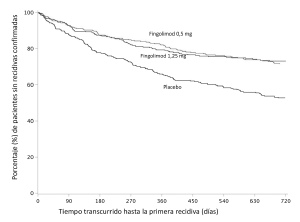
La edad mediana fue de 40,5 años, la duración mediana de la enfermedad, de 8,9 años, y la puntuación mediana de la EDSS al inicio, de 2,5. Los resultados de este estudio se muestran en la tabla 3 y figura 2.
|
Tabla 3. Resultados de la IRM y clínicos del estudio FREEDOMS II |
||
|
GILENYA® 0,5 mg |
Placebo |
|
|
Criterios clínicos |
N=358 |
N=355 |
|
Tasa anualizada de recidivas (criterio principal) |
0,21 (p<0,001*) |
0,40 |
|
Reducción relativa (porcentaje) |
48 |
|
|
Porcentaje de pacientes sin recidivas al cabo de 24 meses |
71,5 (p<0,001*) |
52,7 |
|
Riesgo de progresión de la discapacidad† |
||
|
Cociente de riesgos instantáneos (IC del 95%) (confirmado durante tres meses) |
0,83 (0,61, 1,12) (p=0,227*) |
|
|
Cociente de riesgos instantáneos (IC del 95%) (confirmado durante seis meses) |
0,72 (0,48, 1,07) (p=0,113*) |
|
|
Criterios basados en la IRM |
||
|
Cambio porcentual del volumen cerebral |
n=266 |
n=249 |
|
Cambio porcentual mediano (medio) en 24 meses |
-0,7 (-0,9) (p<0,001*) |
-1,0 (-1,3) |
|
Número de lesiones T2 nuevas o expansivas |
n=264 |
n=251 |
|
Número mediano (medio) en 24 meses |
0,0 (2,3) (p<0,001*) |
4,0 (8,9) |
|
Número de lesiones realzadas con Gd |
n=269 (mes 24) |
n=256 (mes 24) |
|
Número mediano (medio) al |
||
|
Mes 6 |
0,0 (0,2) |
0,0 (1,1) |
|
Mes 12 |
0,0 (0,2) |
0,0 (1,3) |
|
Mes 24 |
0,0 (0,4) (p<0,001* a cada tiempo valorado) |
0,0 (1,2) |
|
Cambio porcentual del volumen total de lesiones en T2 |
n=262 |
n=247 |
|
Cambio porcentual mediano (medio) en 24 meses |
-7,1 (13,7) (p<0,001*) |
0,8 (25,1) |
|
Cambio del volumen de lesiones hipointensas en T1 |
n=225 |
n=209 |
|
Cambio porcentual mediano (medio) en 24 meses |
-9,9 (12,6) (p<0,372) |
-8,5 (26,4) |
Todos los análisis de los criterios clínicos fueron análisis por intención de tratar (análisis IDT). En los análisis de los criterios basados en la IRM se usó un archivo evaluable de datos.
* Indica significación estadística con respecto al placebo con un nivel de confianza bilateral de 0,05.
Determinación de los valores de p: Tasa anualizada de recidivas (TAR) general, por regresión binomial negativa, con ajuste por tratamiento, país, número de recidivas en los 2 años previos y EDSS inicial; porcentaje de pacientes sin recidivas, por el modelo de regresión logística, con ajuste por tratamiento, país, número de recidivas en los 2 años previos y EDSS inicial; tiempo transcurrido hasta la progresión de la discapacidad confirmada a lo largo de 3 o 6 meses, por el modelo de riesgos proporcionales de Cox, con ajuste por tratamiento, país, EDSS inicial y edad; lesiones T2 nuevas o expansivas, por regresión binomial negativa, con ajuste por tratamiento y país; lesiones realzadas con Gd, por análisis de covarianza (ANCOVA) ordinal, con ajuste por tratamiento, país y número inicial de lesiones realzadas con Gd; y cambio porcentual del volumen de lesiones y del volumen cerebral, por ANCOVA ordinal, con ajuste por tratamiento, país y el valor inicial correspondiente.
† Análisis adicionales revelaron que los resultados obtenidos en la población general no eran significativos debido a progresiones falsamente positivas en el subgrupo de pacientes con EDSS inicial =0 (n=62, 8,7% de la población de estudio). En los pacientes con EDSS>0 (n=651; el 91,3% de la población de estudio), la dosis de 0,5 mg de fingolimod produjo una reducción clínicamente importante y estadísticamente significativa en comparación con el placebo (HR= 0,70; IC (0,50, 0,98); p=0,040), igual que en el estudio FREEDOMS.
Figura 3. Gráfico de Kaplan-Meier del tiempo transcurrido hasta la primera recidiva confirmada, hasta el mes 24. Estudio FREEDOMS II (población por IDT)
No se produjeron diferencias significativas en cuanto a los resultados de las variables de evaluación entre la dosis de 0,5 mg y 1,25 mg.
Estudio D2302 (TRANSFORMS): El estudio D2302 (TRANSFORMS) fue un ensayo clínico de fase III, comparativo con tratamiento activo (30 µg de interferón ß-1a administrado por vía intramuscular una vez a la semana), aleatorizado, de doble enmascaramiento y doble simulación y un año de duración, efectuado en pacientes con esclerosis múltiple recidivante remitente que no habían recibido natalizumab en los 6 meses previos. Se permitió la terapia previa con interferón ß o acetato de glatirámero hasta el momento de la aleatorización.
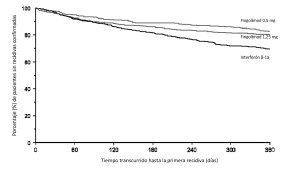
La edad mediana fue de 36 años, la duración mediana de la enfermedad, de 5,9 años, y la puntuación mediana de la EDSS al inicio, de 2,0. Se aleatorizó a los pacientes de forma que recibiesen GILENYA® 0,5 mg (n=431), GILENYA® 1,25 mg (n=426) o interferón ß-1a en dosis de 30 µg por vía intramuscular una vez a la semana (n=435) durante 12 meses. La duración mediana de tratamiento fue de 365 días con GILENYA® 0,5 mg, de 354 días con GILENYA® 1,25 mg y de 361 días con interferón ß-1a.
Los resultados de este estudio se presentan en la Tabla 4 y la Figura 3.
|
Tabla 4. Resultados de la IRM y clínicos del estudio TRANSFORMS |
||
|
GILENYA® 0,5 mg |
Interferón ß-1a, 30 µg, i.m. |
|
|
Criterios clínicos |
N=429 |
N=431 |
|
Tasa anualizada de recidivas (criterio principal) |
0,16 (p<0,001*) |
0,33 |
|
Reducción relativa (porcentual) |
52 |
|
|
Porcentaje de pacientes sin recidivas al cabo de 12 meses |
82,5 (p<0,001*) |
70,1 |
|
Riesgo de progresión de la discapacidad |
||
|
Cociente de riesgos instantáneos (IC del 95%) (confirmado durante tres meses) |
0,71 (0,42, 1,21) (p=0,209*) |
|
|
Criterios basados en la IRM |
||
|
Número de lesiones T2 nuevas o expansivas |
n=380 |
n=365 |
|
Número mediano (medio) en 12 meses |
0,0 (1,7) (p<0,004*) |
1,0 (2,6) |
|
Número de lesiones realzadas con Gd |
n=374 |
n=354 |
|
Número mediano (medio) al cabo de 12 meses |
0,0 (0,2) (p<0,001*) |
0,0 (0,5) |
|
Cambio porcentual del volumen cerebral |
n=368 |
n=359 |
|
Cambio porcentual mediano (medio) en 12 meses |
-0,2 (-0,3) (p<0,001*) |
-0,4 (-0,5) |
Todos los análisis de los criterios clínicos fueron análisis por intención de tratar (análisis IDT). En los análisis de los criterios basados en la IRM se usó un archivo evaluable de datos.
* Indica significación estadística con respecto al interferón ß-1a i.m. con un nivel de confianza bilateral de 0,05.
Determinación de los valores de p: Tasa anualizada de recidivas (TAR) general, por regresión binomial negativa, con ajuste por tratamiento, país, número de recidivas en los 2 años previos y EDSS inicial;
Figura 3. Gráfico de Kaplan-Meier del tiempo transcurrido hasta la primera recidiva confirmada, hasta el mes 12. Estudio TRANSFORMS (población por IDT)
No se produjeron diferencias significativas en cuanto a los resultados de las variables de evaluación entre la dosis de 0,5 mg y 1,25 mg.
Los pacientes que completaron el estudio TRANSFORMS (D2302) tenían la posibilidad de participar en la ampliación del mismo (D2302E1) con doble enmascaramiento. Así pues, 1030 pacientes del estudio principal pasaron a la fase de ampliación y recibieron fingolimod (n=357 continuaron en el grupo de 0,5 mg, 330 siguieron en el grupo de 1,25 mg, 167 del grupo del interferón ß-1a pasaron a recibir 0,5 mg de fingolimod y 176 del grupo del interferón ß-1a pasaron a recibir 1,25 mg de fingolimod). En 882 de estos pacientes (el 85,9%), el seguimiento fue de por lo menos 12 meses durante la fase de ampliación.
Al mes 12 de la ampliación del estudio, los pacientes que en el estudio principal habían recibido interferón ß-1a presentaban reducciones de la TAR del 30% tras su pase al grupo de fingolimod 0,5 mg (cociente de TAR: 0,70, p=0,06). La TAR de los pacientes que habían recibido fingolimod 0,5 mg en el estudio principal fue baja durante la fase principal y la ampliación del estudio (TAR de 0,18 hasta el mes 24).
Los resultados conjuntos de los estudios D2301 (FREEDOMS) y D2302 (TRANSFORMS) revelan una reducción sistemática de la tasa anualizada de recidivas con GILENYA® (en comparación con el tratamiento de referencia) en los subgrupos definidos por sexo biológico, la edad, la terapia previa de la esclerosis múltiple, la actividad de la enfermedad o el grado de discapacidad al inicio.
DATOS SOBRE TOXICIDAD PRECLÍNICA:
El perfil toxicológico preclínico del fingolimod se evaluó en ratones, ratas, perros y monos. Los principales órganos afectados fueron el sistema linfoide (linfocitopenia y atrofia linfoide), los pulmones (aumento de peso, hipertrofia del músculo liso en la zona de unión bronquioalveolar) y el corazón (efecto cronotrópico negativo, aumento de la tensión arterial, alteraciones perivasculares y degeneración miocárdica) en varias especies; los vasos sanguíneos (vasculopatía) de la rata solamente; y la hipófisis, el estómago anterior, el hígado, las glándulas suprarrenales, la porción gastrointestinal y el sistema nervioso de diversas especies con dosis elevadas únicamente (a menudo asociadas a signos de toxicidad general).
Mutagenicidad y carcinogenicidad: El fingolimod no fue mutágeno en una prueba de Ames, ni en la línea celular de linfoma de ratón L5178Y in vitro. No se observaron efectos clastógenos in vitro en las células de pulmón de hámster chino V79. El fingolimod indujo aberraciones cromosómicas cuantitativas (poliploidía) en concentraciones de 3,7 µg/ml o mayores en las células V79, pero no fue clastógeno en las pruebas de micronúcleos realizadas in vivo en ratones y ratas.
No se observaron signos de carcinogenia en un bioensayo de dos años de duración efectuado en ratas con dosis orales de fingolimod de hasta 2,5 mg/kg (la dosis tolerada máxima), lo cual representa un margen de alrededor de 50 veces tomando como base la exposición sistémica humana (AUC) que se alcanza con la dosis de 0,5 mg. No obstante, en un estudio de dos años de duración en ratones, se apreció una mayor incidencia de linfomas malignos con dosis de 0,25 mg/kg o mayores, lo cual representa un margen de alrededor de 6 veces con respecto al AUC humano de la dosis diaria de 0,5 mg.
Toxicidad reproductiva: El fingolimod no afectó al número de espermatozoides ni a la motilidad de estas células, ni tampoco a la fecundidad de las ratas machos y hembras en dosis de hasta 10 mg/kg (la mayor dosis estudiada), lo cual representa un margen de seguridad de casi 150 veces en comparación con el AUC humano de la dosis diaria de 0,5 mg.
El fingolimod no fue teratógeno en las ratas cuando se administró en dosis de 0,1 mg/kg o mayores. Las malformaciones viscerales más frecuentes en los fetos fueron el tronco arterial persistente y defectos del tabique ventricular. Con dosis de 1 mg/kg o mayores se apreció un aumento de pérdidas postimplantacionales en las ratas, y con la dosis de 3 mg/kg hubo una disminución de fetos viables. El fingolimod no fue teratógeno en el conejo, especie en la que se observó una mayor mortalidad embriofetal con dosis de 1,5 mg/kg o mayores, así como una disminución de fetos viables y un retraso del crecimiento fetal con la dosis de 5 mg/kg.
En las ratas, la supervivencia de las crías de la primera generación (F1) se redujo en el período puerperal temprano cuando se administraron dosis que no eran tóxicas para la progenitora. Sin embargo, la administración de fingolimod no afectó al peso corporal, ni al desarrollo, el comportamiento o la fecundidad de los animales de la generación F1. En un estudio de toxicidad en ratas juveniles, no se observaron otros órganos afectados por efectos tóxicos que los ya descritos en ratas adultas. Las estimulaciones repetidas con hemocianina de lapa californiana (KLH) indicaron una respuesta moderadamente reducida durante el período de tratamiento, pero reacciones inmunitarias perfectamente normales al final del período de recuperación de 8 semanas.
El fingolimod pasa a la leche de los animales tratados durante la lactancia. El fingolimod y sus metabolitos atraviesan la barrera placentaria en conejas preñadas.
ADVERTENCIAS Y PRECAUCIONES:
• A todos los pacientes se les debe realizar un electrocardiograma (ECG) basal y controlar la presión arterial antes de administrar la primera dosis de fingolimod y transcurridas 6 horas de la administración. Se debe monitorizar los signos y síntomas de bradicardia en todos los pacientes durante un período de 6 horas, con el control de la frecuencia cardíaca y de la presión arterial cada hora. Durante este período la monitorización electrocardiográfica continua a tiempo real está recomendada.
• Si a las 6 horas el paciente presenta la frecuencia cardíaca más baja de las observadas desde que se le administró la primera dosis del medicamento, se debe prolongar la monitorización durante al menos 2 horas y hasta que la frecuencia cardiaca aumente de nuevo. Adicionalmente, si después de las 6 horas, la frecuencia cardíaca es <45 lpm, o el ECG muestra la aparición de un bloqueo AV de segundo grado o superior, o un intervalo QTc ≥ 500 mseg, la monitorización se debe prolongar (al menos durante toda la noche), y hasta la resolución de condiciones clínicas. La aparición en cualquier momento de un bloqueo AV de tercer grado también conlleva tener que prolongar la monitorización (al menos durante toda la noche).
• Debido al riesgo de alteraciones del ritmo graves fingolimod no debe utilizarse en pacientes con bloqueo AV de segundo grado Mobitz tipo II o superior, síndrome del seno enfermo, bloqueo cardíaco sinoauricular, una historia de bradicardia sintomática o síncope recurrente, o en pacientes con prolongación significativa de QT (QTc > 470 mseg (mujeres) o > 450 mseg (hombres). Como una bradicardia significativa puede ser mal tolerada en pacientes con cardiopatía isquémica conocida (incluyendo angina de pecho), enfermedad cerebrovascular, antecedentes de infarto de miocardio, insuficiencia cardiaca congestiva, antecedentes de paro cardíaco, hipertensión no controlada o apnea del sueño grave, no debe utilizar fingolimod en estos pacientes. En estos pacientes, solo se debe considerar el tratamiento fingolimod si los beneficios esperados superan los riesgos potenciales. Si se considera el tratamiento, antes del inicio del tratamiento se debe pedir consejo a un cardiólogo para determinar la monitorización más adecuada. Al menos para el inicio del tratamiento se recomienda prolongar la monitorización durante toda la noche.
• Fingolimod no se ha estudiado en pacientes con arritmias que requieren tratamiento con medicamentos antiarrítmicos de clase Ia (por ej. quinidina, disopiramida) o de clase III (por ej. amiodarona, sotalol). Los medicamentos antiarrítmicos de clase Ia y clase III en pacientes con bradicardia se han asociado con casos de taquicardia ventricular tipo torsade de pointes. Como el inicio del tratamiento de fingolimod produce una disminución del ritmo cardíaco, fingolimod no se debe utilizar concomitantemente con estos medicamentos.
• En pacientes que reciben tratamiento concomitante con betabloqueantes antagonistas del canal de calcio que disminuyen el ritmo cardíaco (tales como verapamilo, diltiazem o ivabradina) u otras sustancias que pueden disminuir el ritmo cardiaco (p. ej. digoxina, agentes anticolinesterásicos o pilocarpina), la experiencia con fingolimod es limitada. Dado que el inicio del tratamiento con fingolimod también se asocia con una disminución de la frecuencia cardiaca el uso concomitante de estas sustancias durante el inicio del tratamiento con fingolimod puede asociarse con bradicardia grave y bloqueo cardíaco. El tratamiento con fingolimod no debe iniciarse en pacientes que actualmente están en tratamiento con estas sustancias debido al efecto aditivo potencial sobre el ritmo cardiaco. En estos pacientes, el tratamiento con fingolimod solo se debe considerar si los beneficios esperados superan los riesgos potenciales. Si se considera el tratamiento con fingolimod, antes del inicio del tratamiento se debe pedir consejo a un cardiólogo en relación al cambio a medicamentos que no disminuyan el ritmo cardiaco. Si los medicamentos que disminuyen el ritmo cardiaco no pueden ser interrumpidos, se debe pedir el consejo del cardiólogo para determinar la monitorización adecuada de la primera dosis. Se recomienda prolongar la monitorización durante toda la noche.
• Se recomienda repetir el mismo esquema de monitorización establecido para los pacientes a los que se les administra la primera dosis de fingolimod cuando:
— Aparezca bradiarritmia que precise tratamiento farmacológico tras la administración de la primera dosis de fingolimod. Se recuerda que estos pacientes deberán ser monitorizados en un centro médico al menos durante toda la noche.
— Se interrumpa la administración del medicamento durante al menos un día durante las primeras 2 semanas de tratamiento.
— Se interrumpa la administración del medicamento durante más de 7 días durante las semanas 3ª y 4ª de tratamiento.
— Se interrumpa la administración del medicamento durante más de dos semanas después de transcurrido el primer mes de tratamiento.
Si el tratamiento se interrumpe durante períodos de tiempo inferiores a los anteriormente mencionados, la administración de la siguiente dosis de fingolimod podrá realizarse según el calendario inicialmente establecido.
Bradiarritmia: El inicio del tratamiento con GILENYA® produce una disminución transitoria de la frecuencia cardíaca y también puede estar asociado con el retardo de la conducción aurículoventricular (ver Reacciones adversas y Farmacodinámica). La disminución de la frecuencia cardíaca comienza en la hora posterior a la administración de la primera dosis y alcanza su punto máximo en el curso de 6 horas, en la mayoría de los pacientes dentro de las 24 horas. Por esta razón todos los pacientes deben ser monitoreados para los síntomas de bradicardia al menos las 6 primeras horas después de la primera dosis de GILENYA®.
Con la administración continua, la frecuencia cardíaca regresa a su valor inicial en el plazo de un mes de tratamiento crónico (véase Frecuencia y ritmo cardíacos del apartado Farmacodinamia). En los pacientes tratados con GILENYA® 0,5 mg, dicha disminución de la frecuencia cardíaca, medida a través del pulso, es de 8 latidos por minuto (8 lpm), en promedio. Rara vez se ha observado una frecuencia cardíaca inferior a 40 latidos por minuto (véase Reacciones adversas). Los pacientes que tuvieron bradicardias solían ser asintomáticos, pero algunos presentaban síntomas leves o moderados, como hipotensión, mareos, cansancio o palpitaciones, que desaparecían en las primeras 24 horas de tratamiento. Si es necesaria, la bradicardia puede ser tratada con administración parenteral de atropina o isoprenalina.
El inicio del tratamiento con GILENYA® se ha asociado a retrasos de la conducción auriculoventricular (AV), casi siempre en forma de bloqueos auriculoventriculares de primer grado (prolongación del intervalo PR en el electrocardiograma). Se han observado bloqueos auriculoventriculares de segundo grado, que por lo general eran bloqueos de Mobitz de tipo I (Wenckebach), en menos del 0,2% de los pacientes que recibieron GILENYA® 0,5 mg en los ensayos clínicos. Los trastornos de la conducción eran usualmente transitorios, asintomáticos, no requerían tratamiento y se resolvían en las primeras 24 horas de tratamiento. Durante el uso comercial de GILENYA® se han descrito casos aislados de bloqueo auriculoventricular completo, transitorio y de resolución espontánea (véase Reacciones adversas y farmacodinamia).
La medición del monitoreo cardiaco de la primera dosis (ver Tabla resumen bajo la sección Dosificación/Administración).
En todos los pacientes, un electrocardiograma de 12 niveles debe ser llevado a cabo antes de la primera dosis y al final del periodo de las 6 horas de monitoreo.
Por consiguiente, al inicio del tratamiento con GILENYA®, se recomienda la observación de todos los pacientes mediante la determinación del pulso y de la presión arterial una vez por hora, durante un período de 6 horas, por si aparecen signos y síntomas de bradicardia. Antes de la administración del medicamento y al final del período de monitorización de 6 horas se debe realizar un electrocardiograma en todos los pacientes. Si aparecen síntomas vinculados a la bradiarritmia posteriores a una dosis, se debe instaurar un tratamiento adecuado, si procede, y someter al paciente a observación hasta que los síntomas desaparezcan. Si un paciente necesita una intervención farmacológica durante el período de observación posterior a la primera dosis, se debe establecer una monitorización nocturna en un centro médico, y la estrategia de monitorización que se aplicó tras la administración de la primera dosis debe repetirse después de la segunda dosis de GILENYA®.
Si a las 6 horas el paciente presenta la frecuencia cardiaca más baja de las observadas desde que se le administró la primera dosis del medicamento (que sugiere que el efecto farmacodinámico máximo sobre el corazón todavía no se ha manifestado), se debe prolongar la monitorización durante al menos 2 horas y hasta que la frecuencia cardiaca aumente de nuevo.
Adicionalmente, el monitoreo cardiaco continuo, al menos una noche, es requerido si uno de los siguientes criterios está presente:
• Bloqueo AV de tercer grado de nueva aparición en cualquier momento durante la fase de seguimiento tras el inicio del tratamiento.
• 6 horas después del inicio del tratamiento, la presencia de:
— Frecuencia cardiaca < 45 latidos por minuto.
— Nuevo bloqueo AV de segundo grado persistente o bloqueo AV de mayor grado.
— Intervalo QTc ≥ 500 msec.
En ciertas poblaciones de pacientes, GILENYA® puede considerarse si el beneficio esperado supere el riesgo potencial. Bradicardia puede ser mal tolerada en pacientes con enfermedad isquémica conocida (incluyendo angina de pecho), antecedentes de infarto de miocardio, enfermedad cerebrovascular, historia de síncope recurrente y/o bradicardia sintomática, hipertensión no controlada o apnea severa del sueño no tratada. Si el tratamiento con GILENYA® está siendo considerada, la advertencia de un cardiólogo debe ser consultada previo al tratamiento al monitoreo cardiaco adecuado (al menos en la noche). Ver Interacciones.
Se tiene escasa experiencia de uso de GILENYA® en pacientes que reciben tratamiento simultáneo con betabloqueantes, antagonistas del calcio capaces de reducir la frecuencia cardíaca (como el verapamilo, el diltiazem o la ivabradina) u otras sustancias que pueden reducir dicha frecuencia (p.ej.: digoxina). Como el inicio del tratamiento con GILENYA® también está asociado a una ralentización de la frecuencia cardíaca (véase Bradiarritmia), el uso concomitante de dichas sustancias al inicio de la terapia con GILENYA® puede asociarse a bradicardia grave y bloqueo auriculoventricular. Debido al posible efecto aditivo sobre la frecuencia cardíaca, por lo general, el tratamiento con GILENYA® no debe instaurarse en pacientes tratados simultáneamente con tales sustancias. Cuando se considere la posibilidad de tratamiento con GILENYA®, se debe pedir asesoramiento a un cardiólogo sobre la alternativa de usar medicamentos que no disminuyan la frecuencia cardíaca o una monitorización adecuada para iniciar el tratamiento. Pacientes en los que el cambio a otro medicamento no es una opción, debe por lo menos ser monitoreados durante la noche a través de la monitorización electrocardiográfica continua, véase Interacciones.
Si la terapia con GILENYA® se interrumpe más de dos semanas después del primer mes de tratamiento, los efectos sobre la frecuencia cardíaca y la conducción auriculoventricular pueden repetirse al reinstaurarla y se deben tomar las mismas precauciones que se tomaron cuando se administró la primera dosis. Dentro de las primeras dos semanas de tratamiento se recomiendan los procedimientos que se aplicaron cuando se administró la primera dosis después de la interrupción del tratamiento por un día o más, durante las semanas 3 y 4 del tratamiento, las mismas medidas de precaución como para la primera dosis son recomendadas después de la interrupción del tratamiento por más de 7 días.
Prolongación QT: La prolongación del intervalo QT ha sido reportado en muchos pacientes expuestos a GILENYA® (pacientes individuales con prolongación QTcF entre 30 y 60 msec; no prolongación QTcF > 60 msec y no resultados individuales > 500 msec). Pacientes con riesgo de prolongación QTc no fueron incluidos en este estudio. La importancia clínica de estos hallazgos no es clara.
Desde el inicio de los resultados del tratamiento GILENYA® en la frecuencia cardíaca disminuida y una prolongación del intervalo QT, GILENYA® está contraindicada en pacientes con una línea basal QTc en el intervalo de 500 msec (ver “Contraindicaciones”). Si el tratamiento con GILENYA® está siendo considerado en los siguientes grupos de pacientes, el asesoramiento de un cardiólogo debe ser consultado para determinar el monitoreo cardiaco adecuado (incluyendo monitoreo continuo ECG por los menos una noche en un centro médico):
• Pacientes con prolongación QTc significativa (QTc > 470 msec en mujeres, QTc > 450 msec en varones) antes del inicio del tratamiento.
• Pacientes con factores de riesgo adicional a la ocurrencia de la prolongación QT (por ejemplo: hipocalemia, hipomagnesemia o síndrome congénito QT mayor) (ver Farmacodinamia e Interacciones).
El monitoreo cardiaco continuado es necesario al menos una noche en pacientes con un intervalo QTc ≥ 500 msec al final de la fase de monitoreo de las 6 horas siguientes a la iniciación del tratamiento (ver Dosificación/Administración).
GILENYA® no ha sido estudiado en pacientes con arritmias requiriendo tratamiento con clase Ia (por ejemplo quinidina, procainamida) or clase III drogas antiarritmicas (por ejemplo amiodarona, sotalol). La clase Ia y clase III drogas antiarritmicas han sido asociadas con, entre otras cosas, casos de torsade de pointes en pacientes con bradicardia. Desde el inicio de los resultados del tratamiento GILENYA® en la frecuencia cardíaca disminuida, GILENYA® no debe ser usada concomitantemente con cada droga (ver Contraindicaciones).
Infecciones: Un efecto farmacodinámico básico de GILENYA® es la reducción dosis dependiente de la cifra de linfocitos periféricos al 20–30% de los valores iniciales a causa del secuestro reversible de linfocitos en los tejidos linfoides (véase Farmacocinética).
Los efectos de GILENYA® sobre el sistema inmunitario (véase Farmacocinética) pueden incrementar el riesgo de infecciones (véase Reacciones adversas). GILENYA® no debe ser iniciado en pacientes con infecciones activas crónicas o agudas (ver Contraindicaciones). Por lo tanto, se deben emplear estrategias diagnósticas y terapéuticas eficaces en pacientes con síntomas de infección durante la terapia, especialmente si la infección con herpes virus es detectada. Después de retirar el tratamiento con GILENYA®, el fingolimod puede tardar hasta dos meses en eliminarse del organismo, de modo que se aconseja seguir vigilando los signos de infección durante este período (véase el subapartado Retirada del tratamiento).
Los antineoplásicos, los inmunomoduladores o los inmunodepresores (como los corticoesteroides) deben coadministrarse con cautela debido al riesgo de efectos aditivos en el sistema inmunitario. Las decisiones específicas sobre la posología y la duración del tratamiento con corticoesteroides deben basarse en el criterio clínico. La coadministración de un tratamiento breve con corticoesteroides (de hasta 5 días de duración de acuerdo con los protocolos de los estudios) no aumentó la tasa general de infecciones en los pacientes tratados con fingolimod de los ensayos clínicos de fase III en comparación con el placebo. Estos datos indican que se puede administrar un tratamiento breve con corticoesteroides (de hasta 5 días) junto con GILENYA® (véanse Reacciones adversas e Interacciones).
Hay que pedir a los pacientes que reciben GILENYA® que comuniquen los síntomas de infección al médico. Si el paciente contrae una infección grave, se debe considerar la posibilidad de suspender el tratamiento con GILENYA® y, antes de reanudarlo, se deben sopesar los riesgos y los beneficios del mismo.
Vacunas: Las vacunas pueden resultar menos eficaces durante el tratamiento con GILENYA® y hasta dos meses después de su retirada (véase el subapartado Retirada del tratamiento). Debe evitarse el uso de vacunas atenuadas elaboradas con microbios vivos (véase Interacciones).
Es necesario evaluar si el paciente presenta anticuerpos contra la varicela antes del tratamiento con GILENYA®. Así pues, antes de iniciar la terapia con GILENYA®, se recomienda efectuar una prueba de detección de anticuerpos contra el virus de la varicela-zóster (VZV) en todos los pacientes que carezcan de antecedentes de varicela confirmados por un profesional sanitario o de documentación que certifique un ciclo completo de vacunación contra la varicela. Si la prueba da resultados negativos, se recomienda someter al paciente a un ciclo completo de vacunación contra la varicela antes de comenzar el tratamiento con GILENYA® (véase Reacciones adversas). El inicio del tratamiento con GILENYA® debe postergarse un mes para permitir que la vacuna surta todo su efecto.
Edema macular: Se han registrado casos de edema macular, con o sin síntomas visuales (véase Reacciones adversas), en el 0,5% de los pacientes tratados con GILENYA® 0,5 mg, principalmente en los primeros 3-4 meses de tratamiento. Por consiguiente, se aconseja una exploración oftalmológica 3-4 meses después de iniciar el tratamiento. Los pacientes que refieren trastornos de la vista, se debe chequear cada 6 meses por el neurólogo tratante. Si los pacientes reportan problemas visuales en algún momento durante el tratamiento con GILENYA®, el examen del fondo de ojo incluyendo la mácula debe ser evaluado. Las evaluaciones regulares oftalmológicas deben ser llevadas a cabo durante el tratamiento con GILENYA® en pacientes con diabetes mellitus o una historia de uveítis y en pacientes con una historia de edema macular (ver Contraindicaciones).
Función hepática: Los valores de las enzimas hepáticas deben ser determinados al inicio del tratamiento con GILENYA® y, al mes 1, 3 y 6 después del inicio del tratamiento. Los valores de las enzimas hepáticas deben ser evaluados periódicamente durante todo el curso del tratamiento, incluso en la ausencia de síntomas clínicos.
Se han registrado cifras elevadas de enzimas hepáticas, principalmente de alaninaaminotransferasa (ALT). En los ensayos clínicos en pacientes con esclerosis múltiple (EM), se registraron cifras de ALT por lo menos tres veces mayores en el 8,0% (placebo 1.9%) de los pacientes del grupo de GILENYA® 0,5 mg. Incremento de 5 veces el LSN ocurrieron en el 1,8% (placebo 0.9%) de los pacientes que recibían fingolimod y el tratamiento fue discontinuado en cada caso. En algunos pacientes, con la reexposición se observó la reaparación del incremento de las transaminasas hepáticas, lo que apoya una relación con fingolimod. Estrecha vigilancia debe llevarse a cabo si los niveles de transaminasas (particularmente ALT) si se demuestra que los niveles de transaminasas aumentan en más de 5 veces el límite superior de la normalidad (LSN). El tratamiento con GILENYA® debe ser interrumpido si se repite la prueba de los niveles de transaminasas (particularmente los niveles de ALT) incrementándose en más de 5 veces que el LSN, y no debe ser reincorporado hasta que los valores hayan retornado a la normalidad.
La ingestión de otros medicamentos/sustancias (incluyendo bebidas alcohólicas) debe ser evitada. Pacientes con cirrosis hepáticas y disfunción hepática (Clase B y C de Child-Pugh) no deben ser tratadas con GILENYA®. Adicionalmente, ningún tratamiento debe ser dado a pacientes con infección de hepatitis B activa crónica o aguda debido al riesgo de exacerbación de la enfermedad hepática viral (ver Contraindicaciones).
En pacientes que desarrollan síntomas sugestivos de disfunción hepática, tales como náuseas de origen desconocido o vómitos, deben realizarse un control de enzimas hepáticas inmediatamente. GILENYA® debe ser discontinuado si el daño hepático significativo es confirmado (ver Transaminasas hepáticas). La reanudación del tratamiento dependerá si se determina o no otra causa de daño hepático y en los beneficios del paciente con la reanudación del tratamiento versus el riesgo de recurrencia de la disfunción hepática.
Síndrome de encefalopatía reversible posterior (SERP): En los ensayos clínicos y desde la comercialización de GILENYA® , se han registrado casos inusuales de un síndrome de encefalopatía reversible posterior (SERP) con la dosis de 0,5 mg (véase Reacciones adversas). Entre los síntomas notificados figuraban cefaleas intensas de inicio súbito, náuseas, vómitos, alteración del estado mental, trastornos visuales y convulsiones. Los síntomas de SERP son normalmente reversibles, pero pueden evolucionar a ictus isquémico o hemorragia cerebral. Un retraso en el diagnóstico y el tratamiento puede dejar secuelas neurológicas permanentes. Ante la sospecha de un SERP, se debe retirar el tratamiento con GILENYA® .
Efectos respiratorios: Con el tratamiento con GILENYA®, en el Mes 1 se observaron leves disminuciones dosis dependientes en los valores del volumen espiratorio forzado (FEV1) y la capacidad de difusión de monóxido de carbono (DLCO), que después permanecieron estables. Después de los 24 meses de tratamiento, la reducción en FEV1 previsto – como un porcentaje del valor basal – fue 2,7% para 0,5 mg de fingolimod y 1.2% para placebo. Para DLCO, las reducciones de la línea basal después de los 24 meses de tratamiento fueron 3.3% para 0,5 mg de fingolimod y 2.7% para placebo. Los cambios en FEV1 parecen reversibles siguiendo a la discontinuación del tratamiento. La información en la reversibilidad de los cambios de DLCO siguiendo la discontinuación del tratamiento son limitados. En estudios clínicos controlados en pacientes EM ocurrió disnea en 5% de estos con 0,5 mg de fingolimod y 4% de estos con placebo. Muchos pacientes terminaron el tratamiento con GILENYA® debido a la disnea inexplicable en la extensión de estudios (no controlados). GILENYA® no ha sido estudiado en pacientes EM con deterioro de la función pulmonar. Los pacientes que presentan síntomas sugestivos de un desorden pulmonar deben ser examinados por un especialista (con evaluación para incluir espirometría y determinación de DLCO).
Neoplasmas cutáneos: Pacientes con riesgos de neoplasmas cutáneos malignos deben ser sometidos a evaluación dermatológica previos al inicio del tratamiento con GILENYA®, y regularmente durante el curso del tratamiento.
Cambios en el conteo de linfocitos: Basado en el mecanismo de acción, 0,5 mg de GILENYA® reduce visiblemente el conteo de linfocitos al 70% del valor de estado estacionario. Los conteos de sangre deben ser llevados regularmente.
Tratamiento previo con inmunodepresores o inmunomoduladores: Cuando se sustituye un tratamiento con interferón ß o acetato de glatirámero por GILENYA® y siempre que los efectos de tales terapias sobre el sistema inmunitario se hayan resuelto (por ejemplo, las citopenias), no es necesario un período de reposo farmacológico.
Debido a la semivida prolongada del natalizumab, si el tratamiento con GILENYA® comienza en los dos o tres meses siguientes a la retirada de natalizumab, puede haber una exposición simultánea a ambos fármacos y, por consiguiente, efectos concomitantes sobre el sistema inmunitario. Por consiguiente, se recomienda efectuar una evaluación cuidadosa del momento adecuado para instaurar el tratamiento con GILENYA®, estudiando cada caso por separado, cuando el natalizumab se sustituya por GILENYA®.
Cuando se remplacen otros inmunodepresores (por ejemplo mitoxantrona), se ha de tener presente la duración del efecto y el modo de acción de tales sustancias al instaurar GILENYA® para evitar los efectos depresores aditivos sobre el sistema inmunitario.
Retirada del tratamiento: Si se ha tomado la decisión de suspender definitivamente el tratamiento con GILENYA®, el médico debe saber que el fingolimod permanece en la sangre y que ejerce efectos farmacodinámicos, como la disminución de la cifra de linfocitos, hasta dos meses después de la última dosis. La cifra de linfocitos suele normalizarse 1-2 meses después de retirar el tratamiento (véase Farmacocinética). La instauración de otras terapias durante este período causará una exposición concomitante al fingolimod. El uso de inmunodepresores inmediatamente después de suspender el tratamiento con GILENYA® puede producir un efecto aditivo en el sistema inmunitario y por eso es necesario tener precaución.
POSOLOGÍA Y ADMINISTRACIÓN:
Población destinataria general: La dosis recomendada de GILENYA® es una cápsula de 0,5 mg administrada por vía oral una vez al día, que se puede tomar con o sin alimentos. Si se omite una dosis, el tratamiento debe proseguir con la dosis siguiente según lo previsto.
Los pacientes que reciben interferón ß o acetato de glatirámero pueden sustituir directamente su tratamiento por GILENYA® mientras no se observen signos de anomalías importantes relacionadas con el tratamiento, como citopenias. Se aconseja cautela a la hora de sustituir un tratamiento con natalizumab por GILENYA®. La duración del efecto de este medicamento debe considerarse con el fin de evitar los efectos inmunosupresores aditivos. (Véase Advertencias y precauciones: Tratamiento previo con inmunodepresores o inmunomoduladores).
Seguimiento después de la primera dosis de GILENYA®: Un ECG de 12 derivaciones debe llevarse a cabo en todos los pacientes antes del inicio del tratamiento y al final de las 6 horas del periodo de mantenimiento. Después de la primera dosis, se recomienda la observación de todos los pacientes mediante la determinación del pulso y de la tensión arterial una vez por hora, durante un período de 6 horas, por si aparecen signos y síntomas de bradicardia y alteraciones de la conducción aurículoventricular. Se deben tomar previsiones para el tratamiento cardiológico de emergencia. Se recomienda el monitoreo en tiempo real del ECG para las primeras 6 horas después la primera dosis de GILENYA® (véase Advertencias y precauciones - Bradiarritmia).
Algunos pacientes requieren monitoreo cardiaco continuo más allá de las primeras 6 horas después del inicio del tratamiento. (Ver Seguimiento después de la primera dosis de GILENYA® – Tabla resumen en esta Sección Advertencias y Precauciones). Adicionalmente, es responsabilidad del médico tratante para decidir en qué medida es necesario el seguimiento de los parámetro vitales/ECG después de las dosis posteriores.
|
Tabla 1. Monitoreo de seguimiento de la primera dosis de GILENYA® – Tabla resumen |
|
|
Todos los pacientes |
|
|
Deben ser monitoreados cada 6 horas para los síntomas de bradicardia y alteraciones de la conducción aurículo ventricular como sigue: Pulso por hora y la medición de la presión arterial. Un electrocardiograma de 12 derivaciones antes de empezar el tratamiento y después de las 6 horas de monitoreo. Provisiones para el tratamiento cardiológico de emergencia. Se recomienda monitoreo continuo ECG. |
|
|
Pacientes con anormalidades en las primeras 6 horas siguientes a la primera dosis |
|
|
En el evento de bradiarritmia sintomática. |
El paciente debe continuar ser monitoreado siguiendo el periodo de 6 horas de monitoreo hasta que los síntomas hayan sido resueltos. |
|
Si la frecuencia cardiaca alcanza el valor más bajo 6 horas después de la primera dosis. |
El monitoreo cardiaco debe ser continuo hasta que la frecuencia cardiaca se haya recuperado, y por un mínimo de 2 horas. |
|
Si uno de los siguientes hallazgos están presentes en las 6 horas del ECG siguientes de la primera dosis: Frecuencia cardiaca < 45 latidos por minuto. Bloqueo AV de segundo grado persistente nuevo o bloqueo AV de más alto grado. Intervalo QTc ≥ 500 mseg. Si el siguiente hallazgo está presente en algún punto en el tiempo durante la fase de monitoreo siguiendo la primera dosis: Bloqueo AV de inicio reciente de tercer grado. |
El monitoreo cardiaco se debe continuar por lo menos durante la noche. |
|
Si el tratamiento farmacológico de los síntomas relacionados es requerido después de la primera dosis, el paciente debe ser monitoreado toda la noche en un centro médico. La estrategia del monitoreo de la primera dosis debe ser repetido para la administración de una segunda dosis. |
|
|
Pacientes con enfermedad cardiaca preexistente. |
|
|
En determinados pacientes, GILENYA® debe ser considerado solo si el beneficio esperado supera el riesgo potencial. |
|
|
En pacientes predispuestos con: Enfermedad cardiaca isquémica conocida (Incluyendo angina pectoris) Falla cardiaca congestiva. Enfermedad cerebrovascular. Hipertensión descontrolada. Apnea del sueño severa no tratada. Así como los pacientes con un historial de las siguientes enfermedades: Infarto de miocardio. Paro cardíaco. Síncope recurrente. Bradicardia sintomática. |
Lo siguiente debe ser llevado a cabo antes de empezar el tratamiento: Se debe realizar el monitoreo cardiaco adecuado (al menos durante la noche). Se debe realizar el monitoreo cardiaco adecuado (al menos durante la noche). |
|
Los pacientes que toman medicamento para bajar el ritmo cardiaco |
|
|
En pacientes con: Betabloqueadores. Bloqueadores del canal de calcio (con un efecto en la disminución de frecuencia cardiaca como verapamilo, diltiazem, ivabradina). Otras sustancias las cuales pueden disminuir la frecuencia cardiaca (por ejemplo: digoxina, inhibidores de la acetilcolinesterasa [AChEl], pilocarpina). |
Lo siguiente debe ser llevado a cabo antes de iniciar el tratamiento: Se debe consultar a un cardiólogo. Para examinar la posibilidad de cambiar a un medicamento que no ralentiza el ritmo cardíaco o retraso de la conducción AV o Si el paciente no puede cambiar una medicación diferente, debe llevarse a cabo el monitoreo cardiaco (incluyendo ECG continuo) por lo menos una noche. |
|
Pacientes con prolongación del intervalo QT |
|
|
En pacientes con: Prolongación QTc significante (QTc > 470 msec en mujeres, QTc > 450 msec en hombres) antes de iniciar el tratamiento. Factores adicionales de riesgo para la ocurrencia de la prolongación QT (por ejemplo: hipocalemia, hipomagnesemia o síndrome de QT largo congénito). |
Lo siguiente debe ser llevado a cabo antes de iniciar el tratamiento: Se debe consultar a un cardiólogo. Se recomienda el monitoreo cardiaco adecuado (incluyendo ECG continuo al menos durante la noche en un centro médico). |
Posología en poblaciones especiales:
• Disfunción renal: No hay información clínica en seguridad y eficacia en pacientes con disfunción renal.
• Disfunción hepática: No es necesario ajustar la dosis de GILENYA® en pacientes con disfunción hepática leve (clase A de Child-Pugh) debe utilizarse con cautela cuando se trata a estos pacientes (ver Advertencias y Precauciones, función hepática y Farmacocinética). GILENYA® no debe utilizarse en pacientes con disfunción hepática moderada (clase B de Child-Pugh) o disfunción hepática grave (clase C de Child-Pugh) (véase Contraindicaciones).
• Pacientes pediátricos y adolescentes: GILENYA® no está indicado para uso pediátrico ni en adolescentes (véase Farmacocinética y contraindicaciones).
• Pacientes geriátricos: Información clínica para pacientes con esclerosis múltiple mayores de 55 años de edad es muy limitada.
• Origen étnico: No es necesario ajustar la dosis de GILENYA® según el origen étnico de la persona (véase Farmacocinética).
• Sexo biológico: No es necesario ajustar la dosis de GILENYA® según el sexo de la persona (véase Farmacocinética).
INSTRUCCIONES DE USO Y MANIPULACIÓN:
Ningún requisito en especial.
Fabricante: Novartis Pharma Stein AG - Suiza
Revisión de texto: Septiembre de 2013
NOVARTIS PHARMA AG, Basilea (Suiza)
® Marca registrada
SOBREDOSIS: Dosis únicas hasta 80 veces mayores que la dosis recomendada (de 0,5 mg) fueron bien toleradas en voluntarios sanos. Con la dosis de 40 mg, 5 o 6 personas refirieron una ligera opresión o malestar torácicos que eran clínicamente indicativos de una leve reactividad de las vías respiratorias.
El fingolimod puede inducir bradicardia y causar conducción auriculoventricular lento. El descenso de la frecuencia cardíaca suele comenzar durante la hora posterior a la administración de la primera dosis y alcanza su valor máximo en un plazo de 6 horas después de la administración. Ha habido informes de conducción auriculoventricular lenta y comunicaciones aisladas de un bloqueo auriculoventricular completo, de carácter transitorio y resolución espontánea (véanse Advertencias y precauciones y Reacciones adversas).
En caso de sobredosis en un paciente que recibe GILENYA® es importante vigilar la aparición de signos y síntomas de bradicardia y bradiarritmia. Si la sobredosis ocurre al inicio del tratamiento, es importante monitorear a los pacientes con el seguimiento ECG continuo y determinar el pulso por hora y la medida de la presión arterial por al menos las primeras 6 horas. Las mismas medidas como el monitoreo en las primeras dosis aplican (ver Tabla 1: Monitoreo de seguimiento de la primera dosis de GILENYA® en la sección Dosificación/Administración). Ni la diálisis ni la plasmaféresis lograrán eliminar significativamente al fingolimod del organismo.
FARMACOLOGÍA CLÍNICA:
Propiedades/Acción: Código ATC: L04AA27
Modo de acción: El fingolimod es un modulador de los receptores de la esfingosina-1-fosfato. La enzima esfingosina-cinasa lo metaboliza y convierte en el metabolito activo fosfato de fingolimod (fingolimod-fosfato). El fingolimod-fosfato se fija (en concentraciones nanomolares ínfimas) a los receptores 1, 3 y 4 de la esfingosina-1-fosfato (S1P) localizados en los linfocitos y atraviesa fácilmente la barrera hematoencefálica para fijarse a los receptores 1, 3 y 5 de la S1P presentes en las células neurales del sistema nervioso central. Al actuar como antagonista funcional de los receptores de la S1P en los linfocitos, el fingolimod-fosfato bloquea la capacidad de estos últimos para egresar de los ganglios linfáticos y causa de ese modo una redistribución de dichas células y no su disminución. Esta redistribución reduce la infiltración de linfocitos patógenos (como las células Th17 proinflamatorias) en el interior del sistema nervioso central, donde hubieran participado en la inflamación y el daño del tejido nervioso.
Los estudios en animales y los experimentos in vitro indican que el fingolimod también puede ejercer un efecto beneficioso en la esclerosis múltiple gracias a su interacción con los receptores de la S1P en las células neurales. El fingolimod penetra en el SNC de los seres humanos y animales y reduce la astrogliosis, la desmielinización y la pérdida neuronal.
Además, el tratamiento con fingolimod aumenta la concentración de BDNF (factor neurótropo derivado del cerebro) en la corteza, el hipocampo y el cuerpo estriado del cerebro, lo cual favorece la supervivencia neuronal y mejora las funciones motoras.
Propiedades farmacodinámicas:
• Trastornos del sistema inmunitario:
Efectos sobre las cifras de las células inmunitarias de la sangre. Entre 4 y 6 horas después de la administración de la primera dosis de 0,5 mg de fingolimod, la cifra de linfocitos desciende hasta cerca del 75% del valor inicial. Dicha cifra continúa disminuyendo a lo largo de dos semanas con la administración diaria continua hasta alcanzar su nivel mínimo de casi 500 células/µl o aproximadamente el 30% de la cifra inicial. El 18% de los pacientes presentan mínimos por debajo de 200 células/µl en al menos una ocasión. La cifra baja de linfocitos persiste con la administración diaria crónica. La mayoría de los linfocitos T y B circulan habitualmente entre los órganos linfoides, y el fingolimod afecta principalmente a estas células. Entre el 15 y el 20% de los linfocitos T son de fenotipo efector y de memoria, es decir, células que desempeñan una función importante en la vigilancia inmunitaria periférica.
Este subgrupo de linfocitos normalmente no se traslada a los órganos linfoides y por ese motivo no es afectado por el fingolimod. El número de linfocitos periféricos aumenta a los pocos días de retirar el tratamiento con GILENYA® y generalmente se normaliza en un plazo de uno o dos meses. La administración crónica de fingolimod reduce levemente el número de neutrófilos a cerca del 80% del valor inicial. El fingolimod no afecta a los monocitos.
• Frecuencia y ritmo cardíacos: El fingolimod reduce transitoriamente la frecuencia cardíaca y la conducción auriculoventricular al inicio del tratamiento (véase Reacciones adversas). El descenso máximo de la frecuencia cardíaca se observa en las 6 horas siguientes a la administración, y el 70% del efecto cronotrópico negativo se manifiesta el primer día. La frecuencia cardíaca regresa progresivamente a su nivel inicial en el plazo de un mes de tratamiento crónico.
El tratamiento con fingolimod no afecta las respuestas autónomas del corazón, como la variación diurna de la frecuencia cardíaca y la respuesta al ejercicio.
Al inicio del tratamiento con fingolimod, se observa un aumento de extrasístoles auriculares, pero no una mayor frecuencia de fibrilación o aleteo auriculares ni de arritmias o de ectopias ventriculares. El tratamiento con fingolimod no se asocia a una disminución del gasto cardíaco.
La atropina, la isoprenalina o el salmeterol pueden revertir la disminución de la frecuencia cardíaca inducida por el fingolimod.
• Potencial de prolongación del intervalo QT: En un estudio exhaustivo del intervalo QT con dosis de 1,25 o de 2,5 mg de fingolimod, en el estado estacionario, cuando el fármaco todavía ejercía un efecto cronotrópico negativo, se observó una prolongación del intervalo QTcI con un límite superior del intervalo de confianza del 90% ≤ 13,0 ms. No existe relación entre la dosis (o la exposición) y el efecto del fingolimod y la prolongación del QTcI. El tratamiento con fingolimod no se asoció a una señal persistente de aumento de la incidencia de valores atípicos del QTcI, ya fuese éste absoluto o relativo con respecto al inicio. Sin embargo, en el estudio 1, la prolongación QTcF entre 30 y 60 msec ocurrió después de la dosis inicial de 0,5 mg de fingolimod en 6,6% de pacientes (placebo: 2.4%) y en 13.9% (placebo: 6,7%) de pacientes durante el curso completo del tratamiento. La relevancia clínica es desconocida.
• Función pulmonar: El tratamiento con dosis únicas o múltiples de 0,5 y 1,25 mg de fingolimod de dos semanas de duración no se asocia a un aumento detectable de la resistencia de las vías respiratorias (al paso del aire), medida a través del FEV1 y del flujo espiratorio forzado entre el 25 y el 75% de la capacidad vital forzada (FEF25-75). Sin embargo, dosis únicas de fingolimod ≥5 mg (diez veces de la dosis recomendada) se asocian a un aumento de la resistencia de las vías respiratorias que depende de la dosis. El tratamiento con dosis múltiples de 0,5, 1,25 o 5 mg de fingolimod no se asocia a una oxigenación insuficiente ni a una desaturación de oxígeno con el ejercicio, ni tampoco a una mayor sensibilidad de las vías respiratorias a la metacolina. Los sujetos en tratamiento con fingolimod presentan una respuesta broncodilatadora normal frente a los agonistas ß inhalados.
Propiedades farmacocinéticas:
Absorción: La absorción del fingolimod es lenta (tmáx de 12-16 horas) y considerable (85%; el cálculo se basa en la cantidad de radioactividad eliminada en la orina y en la cantidad de metabolitos fecales extrapolada al infinito). Su aparente biodisponibilidad oral absoluta es alta (93%).
La ingestión de alimentos no altera la Cmáx del fingolimod o del fingolimod-fosfato ni la exposición (AUC) a dichas sustancias. Por consiguiente, GILENYA® se puede administrar con independencia del horario de la comida (véase Posología y administración).
Las concentraciones sanguíneas estacionarias se alcanzan en 1 o 2 meses con la pauta de administración de una sola vez al día, y las concentraciones estacionarias son unas diez veces mayores que las que se obtienen con la dosis inicial.
Distribución: El fingolimod se distribuye ampliamente entre los eritrocitos, y la fracción absorbida en las células sanguíneas es del 86%. La absorción del fingolimod-fosfato en dichas células es menor <17%. El fingolimod y el fingolimod-fosfato se hallan principalmente unidos a proteínas (>99,7%). La disfunción renal o hepática no altera la unión proteínica del fingolimod o del fingolimod-fosfato.
Fingolimod se distribuye ampliamente entre los tejidos del cuerpo, con un volumen de distribución de 1200±260 l. Un estudio en cuatro sujetos sanos que recibieron una dosis intravenosa única de fingolimod marcado con un isótopo del iodo demostró que el fingolimod ingresa en el cerebro. En un estudio de 13 pacientes varones con esclerosis múltiple que recibieron 0,5 mg diarios de GILENYA® se observó que, en el estado estacionario, la cantidad de fingolimod (y de fingolimod-fosfato) en el semen era más de 10 000 veces menor que la dosis administrada (0,5 mg).
Metabolismo: La biotransformación del fingolimod en el ser humano ocurre básicamente por tres vías: Por fosforilación estereoselectiva y reversible al metabolito farmacológicamente activo, el (S)-enantiómero del fosfato de fingolimod, por biotransformación oxidativa catalizada principalmente por el CYP4F2 (y posiblemente por otras isoformas del CYP4F) y la consiguiente degradación de tipo cetoácida a metabolitos inactivos, y por formación de análogos del fingolimod ceramídicos, no polares y farmacológicamente inactivos.
Tras la administración única de 14C-fingolimod por vía oral, los principales componentes fingolimódicos de la sangre –a juzgar por su contribución al AUC del total de componentes radioactivos hasta 816 horas después de la administración– son el propio fingolimod (23,3%), el fingolimod-fosfato (10,3%) y metabolitos inactivos como el ácido carboxílico M3 (8,3%), el cerámido M29 (8,9%) y el cerámido M30 (7,3%).
Eliminación: La depuración sanguínea del fingolimod es de 6,3±2,3 l/h y la semivida de eliminación terminal aparente (t½), de 6-9 días, en promedio. La concentración sanguínea de fingolimodfosfato disminuye paralelamente a la de fingolimod en la fase terminal dando por resultado semividas similares para ambos.
Tras la administración oral, cerca del 81% de la dosis se excreta lentamente en la orina en forma de metabolitos inactivos. El fingolimod y el fingolimod-fosfato no se excretan inalterados en la orina, pero son los componentes principales de las heces, donde representan menos del 2,5% de la dosis en cada caso. El 89% de la dosis administrada se recupera al cabo de 34 días.
Linealidad: La concentración de fingolimod y de fingolimod-fosfato aumenta de forma aparentemente proporcional a la dosis después de la administración repetida de 0,5 mg o 1,25 mg de fingolimod una vez al día.
Poblaciones especiales:
• Pacientes con disfunción renal: La disfunción renal grave incrementa la Cmáx y el AUC del fingolimod en un 32% y un 43%, respectivamente, y la Cmáx y el AUC del fingolimod-fosfato en un 25% y un 14%, respectivamente. La semivida de eliminación aparente de ambos análitos permanece inalterada. No es necesario ajustar la dosis de GILENYA® en pacientes con disfunción renal.
• Pacientes con disfunción hepática: La farmacocinética del fingolimod en dosis únicas (de 1 o 5 mg), cuando se evaluó en sujetos con disfunción hepática leve, moderada o grave (clases A, B y C de Child-Pugh), no reveló ninguna alteración de la Cmáx, pero sí un aumento del AUC del 12%, 44% y 103%, respectivamente. La semivida de eliminación aparente permanece inalterada en la disfunción hepática leve, pero se prolonga en un 49-50% en la disfunción hepática moderada o grave. En pacientes con disfunción hepática grave (clase C de Child-Pugh), la Cmáx de fingolimodfosfato es un 22% menor y el AUC, un 38% mayor. No se evaluó la farmacocinética del fingolimod-fosfato en pacientes con disfunción hepática leve o moderada.
• Pacientes pediátricos y adolescentes: No se ha estudiado la inocuidad ni la eficacia de GILENYA® en menores de 18 años de edad.
GILENYA® no está indicado para uso pediátrico.
• Pacientes geriátricos: El modo de eliminación y los resultados de un análisis farmacocinético poblacional indican que no es necesario ajustar la dosis en los pacientes de edad avanzada. No obstante, no se tiene suficiente experiencia clínica en pacientes mayores de 55 años.
• Origen étnico: El origen étnico no ejerce efectos clínicamente importantes en la farmacocinética del fingolimod y el fosfato de fingolimod.
• Sexo biológico: El sexo biológico no afecta en absoluto la farmacocinética del fingolimod o del fosfato de fingolimod.
CONSERVACIÓN: Consérvese a menos de 30 °C.
Debe protegerse de la humedad.
GILENYA® no debe utilizarse después de la fecha de caducidad que figura en el envase («EXP»).
GILENYA® debe conservarse fuera del alcance y de la vista de los niños.
VIDA ÚTIL: 18 meses


