

IMBRUVICA
IBRUTINIB
Cápsulas
Caja, Blíster, Cápsulas, 140 Miligramos
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA:
Cada CÁPSULA contiene:
Ibrutinib 140 mg
Excipientes c.s.p.
Consulte la lista completa de los excipientes en la sección Lista de excipientes.
INDICACIONES Y USO:
Linfoma de células del manto: IMBRUVICA® está indicado para el tratamiento de pacientes con linfoma de células del manto (LCM) quienes han recibido al menos un tratamiento previo.
La aprobación acelerada fue concedida para esta indicación basada en la tasa de respuesta global. La aprobación continua para esta indicación puede estar supeditada a la verificación y descripción del beneficio clínico en un estudio confirmatorio (ver sección Estudios clínicos).
Leucemia linfocítica crónica/Linfoma linfocítico de células pequeñas: IMBRUVICA® está indicado para el tratamiento de pacientes con leucemia linfocítica crónica (LLC)/linfoma linfocítico de células pequeñas (LLCP) (ver sección Estudios clínicos).
Leucemia linfocítica crónica/Linfoma linfocítico de células pequeñas con deleción del 17p: IMBRUVICA® está indicado para el tratamiento de pacientes con leucemia linfocítica crónica (LLC)/Linfoma linfocítico de células pequeñas (LLCP) con deleción del 17p (ver sección Estudios clínicos).
Macroglobulinemia de Waldenström: IMBRUVICA® está indicado para el tratamiento de pacientes con macroglobulinemia de Waldenström (MW) (ver sección Estudios clínicos).
Linfoma de zona marginal: IMBRUVICA® está indicado para el tratamiento de pacientes con linfoma de zona marginal (LZM) que requieren terapia sistémica y que han recibido al menos una terapia previa basada en anti-CD20.
La aprobación acelerada fue concedida para esta indicación basada en la tasa de respuesta global (ver sección Estudios clínicos). La aprobación continua para esta indicación puede estar supeditada a la verificación y descripción del beneficio clínico en un estudio confirmatorio.
DATOS FARMACÉUTICOS:
Lista de excipientes: Las cápsulas de IMBRUVICA® contienen los siguientes excipientes: croscarmelosa de sodio, estearato de magnesio, celulosa microcristalina y lauril sulfato de sodio.
Incompatibilidades: Ninguna reportada.
Período de validez: 24 meses
Observe la fecha de caducidad en el embalaje exterior. No utilice este medicamento después de la fecha de caducidad que aparece en el envase después de “EXPIRA”. La fecha de caducidad es el último día del mes que se indica.
Condiciones de almacenamiento
No conservar a más de 30 °C.
Mantenga fuera de la vista y del alcance de los niños.
Instrucciones para la eliminación: Cualquier producto no utilizado o material de desecho debe eliminarse de acuerdo con las normas locales.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que ya no necesita. De esta forma, ayudará a proteger el medio ambiente.
Titular del registro sanitario:
JOHNSON & JOHNSON DEL PERU S.A.
Av. Canaval y Moreyra N° 480, Int. 901 y 1301
San Isidro-Lima
Desarrollado en conjunto con Pharmacyclics
© Pharmacyclics 2014
© Janssen-Cilag 2014
CONTRAINDICACIONES:
Ninguna.
INFORMACIÓN DE RECOMENDACIÓN A LOS PACIENTES:
Recomendar al paciente leer el Inserto del paciente que se proporciona dentro del envase externo del medicamento.
• Hemorragia: Informar a los pacientes sobre la posibilidad de sangrado y reportar cualquier signo o síntomas (dolor de cabeza severa, sangre en las heces u orina, sangrado prolongado o no controlado). Informar al paciente que IMBRUVICA® puede necesitar interrumpirse por procedimientos médicos o dentales (Ver sección Advertencias y precauciones).
• Infecciones: Informar a los pacientes sobre la posibilidad de infección grave, y reportar cualquier signo o síntoma (fiebre, escalofrío, debilidad, confusión) que sugieran infección (Ver sección Advertencias y precauciones).
• Fibrilación auricular: Recomendar a los pacientes reportar cualquier signo de palpitaciones, aturdimiento, mareos, desmayos, respiración entrecortada y malestar de pecho (Ver sección Advertencias y precauciones).
• Hipertensión: Informar a los pacientes que se han ocurrido presión sanguínea elevada en pacientes que toman IMBRUVICA®, que puede requerir tratamiento con terapia antihipertensiva (Ver sección Advertencias y precauciones).
• Segundas neoplasias malignas: Informar a los pacientes que han ocurrido otras neoplasias malignas en pacientes que han sido tratados con IMBRUVICA®, incluyendo cánceres de piel y otros carcinomas (Ver sección Advertencias y precauciones)
• Síndrome de lisis tumoral: Informar a los pacientes del riesgo potencial de síndrome de lisis tumoral y reportar cualquier signo y síntoma asociado con este evento al profesional de la salud para su evaluación (ver sección Advertencias y precauciones).
• Toxicidad embriofetal: Advertir a las mujeres del riesgo potencial para el feto y evitar el embarazo durante el tratamiento y durante 1 mes después de la última dosis de IMBRUVICA® (ver sección Advertencias y precauciones)
• Informar a los pacientes tomar IMBRUVICA® por vía oral una vez al día de acuerdo a las instrucciones de su médico y que las cápsulas deben ser ingeridas enteras con un vaso de agua, sin abrirlas, romperlas o masticarlas, aproximadamente a la misma hora del día (ver sección Posología y administración)
• Advertir a los pacientes que en caso de omisión de una dosis diaria de IMBRUVICA®, esta debe ser tomada tan pronto como sea posible el mismo día y regresar al esquema normal al día siguiente. Los pacientes no deben tomar cápsulas adicionales para recuperar la dosis omitida (ver sección Posología y administración).
• Advertir a los pacientes sobre los efectos secundarios comunes asociados con IMBRUVICA® (ver sección Reacciones adversas). Dirigir al paciente a una lista completa de las reacciones adversas al fármaco en el INSERTO PARA EL PACIENTE.
• Advertir a los pacientes de informar al profesional de salud sobre todos los medicamentos concomitantes, incluyendo medicamentos de prescripción, sin prescripción, vitaminas y productos a base de hierbas (ver sección Interacciones con medicamentos).
• Advertir a los pacientes que pueden experimentar heces blandas o diarrea y que deben contactar a su médico si la diarrea persiste. Recomendar al paciente mantener hidratación adecuada (ver sección Reacciones adversas).
REACCIONES ADVERSAS:
Las siguientes reacciones adversas se discuten con mayor detalle en otras secciones de la ficha técnica:
• Hemorragia (ver sección Advertencias y precauciones)
• Infecciones (ver sección Advertencias y precauciones)
• Citopenias (ver sección Advertencias y precauciones)
• Fibrilación auricular (ver sección Advertencias y precauciones)
• Hipertensión (ver sección Advertencias y precauciones)
• Segundas neoplasias primarias (ver sección Advertencias y precauciones)
• Síndrome de lisis tumoral (ver sección Advertencias y precauciones).
Experiencia en los estudios clínicos: Debido a que los estudios clínicos son conducidos bajo condiciones ampliamente variables, las tasas de los eventos adversos observados en los estudios clínicos de un fármaco no pueden ser directamente comparados con las tasas de estudios clínicos de otros fármacos y pueden no reflejar las tasas observadas en la práctica.
Linfoma de células del manto: Los datos descritos a continuación reflejan la exposición a IMBRUVICA® en un estudio clínico que incluyó a 111 pacientes con LCM previamente tratados con 560 mg diarios con una mediana de la duración del tratamiento de 8.3 meses.
Las reacciones adversas que ocurrieron comúnmente (≥ 20%) fueron trombocitopenia, diarrea, neutropenia, anemia, fatiga, dolor musculoesquelético, edema periférico, infección del tracto respiratorio superior, náuseas, hematomas, disnea, estreñimiento, erupción, dolor abdominal, vómitos y disminución del apetito (ver Tablas 1 y 2).
Las reacciones adversas no hematológicas de grado 3 o 4 (≥ 5%) más comunes fueron neumonía, dolor abdominal, fibrilación auricular, diarrea, fatiga e infecciones de la piel.
Casos de insuficiencia renal fatal y grave han ocurrido con la terapia con IMBRUVICA®. Incrementos de la creatinina en 1.5 a 3 veces el límite superior normal ocurrieron en el 9% de los pacientes.
Las reacciones adversas del estudio de LCM (N = 111) usando 560 mg diarios de IMBRUVICA® en monoterapia, que ocurrieron con una tasa ≥ 10% se presentan en la Tabla 1.
|
Tabla 1: Reacciones adversas no hematológicas informadas en ≥ 10% de los pacientes con LCM (N=111) |
|||
|
Sistema del cuerpo |
Reacción Adversa |
Todos los grados (%) |
Grados 3 o 4 (%) |
|
Trastornos gastrointestinales |
Diarrea |
51 |
5 |
|
Náuseas |
31 |
0 |
|
|
Estreñimiento |
25 |
0 |
|
|
Dolor abdominal |
24 |
5 |
|
|
Vómito |
23 |
0 |
|
|
Estomatitis |
17 |
1 |
|
|
Dispepsia |
11 |
0 |
|
|
Infecciones e infestaciones |
Infección del tracto respiratorio superior |
34 |
0 |
|
Infección del tracto urinario |
14 |
3 |
|
|
Neumonía |
14 |
7 |
|
|
Infecciones de la piel |
14 |
5 |
|
|
Sinusitis |
13 |
1 |
|
|
Trastornos generales y condiciones en el lugar de la administración |
Fatiga |
41 |
5 |
|
Edema periférico |
35 |
3 |
|
|
Pirexia |
18 |
1 |
|
|
Astenia |
14 |
3 |
|
|
Trastornos de la piel y del tejido subcutáneo |
Hematoma |
30 |
0 |
|
Erupción |
25 |
3 |
|
|
Petequias |
11 |
0 |
|
|
Trastornos músculo- esqueléticos y del tejido conectivo |
Dolor musculoesquelético |
37 |
1 |
|
Espasmos musculares |
14 |
0 |
|
|
Artralgia |
11 |
0 |
|
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
27 |
4 |
|
Tos |
19 |
0 |
|
|
Epistaxis |
11 |
0 |
|
|
Trastornos del metabolismo y la nutrición |
Disminución del apetito |
21 |
2 |
|
Deshidratación |
12 |
4 |
|
|
Trastornos del sistema nervioso |
Mareos |
14 |
0 |
|
Dolor de cabeza |
13 |
0 |
|
|
Tabla 2: Disminución de la hemoglobina, plaquetas o neutrófilos emergente* del tratamiento en pacientes con LCM (N=111) |
||
|
|
Porcentaje de Pacientes (N = 111) |
|
|
Todos lo grados (%) |
Grado 3 o 4 (%) |
|
|
Disminución de plaquetas |
57 |
17 |
|
Disminución de neutrófilos |
47 |
29 |
|
Disminución de hemoglobina |
41 |
9 |
|
*En base a las medidas de laboratorio y las reacciones adversas |
||
Diez pacientes (9%) discontinuaron el tratamiento debido a las reacciones adversas en el estudio (N = 111). La reacción adversa más frecuente que condujo a la discontinuación del tratamiento fue hematoma subdural (1.8%).
Las reacciones adversas que condujeron a la reducción de la dosis ocurrieron en el 14% de los pacientes.
Los pacientes con LCM que desarrollaron linfocitosis mayor a 400 000/mcL han desarrollado hemorragia intracraneal, letargia, inestabilidad de la marcha y dolor de cabeza. Sin embargo, algunos de estos casos se encontraban en el contexto de progresión de la enfermedad.
El cuarenta por ciento de los pacientes tuvieron niveles elevados de ácido úrico en el estudio incluyendo el 13% con valores por encima de 10 mg/dL. La reacción adversa de hiperuricemia fue reportada por el 15% de los pacientes.
Leucemia linfocítica crónica/Linfoma linfocítico de células pequeñas: Los datos descritos a continuación reflejan la exposición en un estudio clínico abierto de un solo brazo y tres estudios clínicos aleatorizados, controlados, en pacientes con LLC/LLCP (n = 1278 total y n = 668 pacientes expuestos a IMBRUVICA®). El Estudio 1 incluyó 51 pacientes con LLC/LLCP previamente tratados, el Estudio 2 incluyó 391 pacientes aleatorizados con LLC o LLCP previamente tratados quienes recibieron IMBRUVICA® u ofatumumab en monoterapia, el Estudio 3 incluyó 269 pacientes aleatorizados de 65 años o mayores con LLC o LLCP sin tratamiento previo quienes recibieron IMBRUVICA® o clorambucil en monoterapia y el Estudio 4 incluyó 578 pacientes aleatorizados con LLC o LLCP previamente tratados quienes recibieron IMBRUVICA® en combinación con bendamustina y rituximab o placebo en combinación con bendamustina y rituximab.
Las reacciones adversas más comunes que ocurrieron en los Estudios 1, 2, 3 y 4 en los pacientes con LLC/LLCP que recibieron IMBRUVICA® (≥ 20%) fueron neutropenia, trombocitopenia, anemia, diarrea, dolor musculoesquelético, náuseas, erupción, hematomas, fatiga, pirexia y hemorragia. Cuatro al 10% de los pacientes que recibieron IMBRUVICA® en el Estudio 1, 2, 3 y 4 discontinuaron el tratamiento debido a las reacciones adversas. Estas incluyeron neumonía, hemorragia, fibrilación auricular, erupción y neutropenia (1% de cada uno). Las reacciones adversas que condujeron a la reducción de la dosis ocurrieron aproximadamente en el 6% de los pacientes.
Estudio 1: Las reacciones adversas y anormalidades de laboratorio del estudio de LLC/LLCP (N=51) usando 420 mg al día de IMBRUVICA® en monoterapia en pacientes con LLC/LLCP previamente tratados que ocurrieron a una tasa ≥ 10 % con una mediana de la duración de tratamiento de 15.6 meses se presentan en la Tabla 3 y la Tabla 4.
|
Tabla 3: Reacciones adversas no hematológicas en ≥ 10% de los pacientes con LLC/LLCP (N=51) en el Estudio 1 |
|||
|
Sistema del cuerpo |
Reacción Adversa |
Todos los grados (%) |
Grados 3 o 4 (%) |
|
Trastornos gastrointestinales |
Diarrea |
59 |
4 |
|
Estreñimiento |
22 |
2 |
|
|
Náuseas |
20 |
2 |
|
|
Estomatitis |
20 |
0 |
|
|
Vómitos |
18 |
2 |
|
|
Dolor abdominal |
14 |
0 |
|
|
Dispepsia |
12 |
0 |
|
|
Infecciones e infestaciones |
Infección del tracto respiratorio superior |
47 |
2 |
|
Sinusitis |
22 |
6 |
|
|
Infección de la piel |
16 |
6 |
|
|
Neumonía |
12 |
10 |
|
|
Infección del tracto urinario |
12 |
2 |
|
|
Trastornos generales y condiciones en el lugar de la administración |
Fatiga |
33 |
6 |
|
Pirexia |
24 |
2 |
|
|
Edema periférico |
22 |
0 |
|
|
Astenia |
14 |
6 |
|
|
Escalofríos |
12 |
0 |
|
|
Trastornos de la piel y del tejido subcutáneo |
Hematoma |
51 |
2 |
|
Erupción |
25 |
0 |
|
|
Petequias |
16 |
0 |
|
|
Trastornos respiratorios, torácicos y mediastínicos |
Tos |
22 |
0 |
|
Dolor orofaríngeo |
14 |
0 |
|
|
Disnea |
12 |
0 |
|
|
Trastornos musculoesqueléticos y del tejido conectivo |
Dolor musculoesquelético |
25 |
6 |
|
Artralgia |
24 |
0 |
|
|
Espasmos musculares |
18 |
2 |
|
|
Trastornos del sistema nervioso |
Mareos |
20 |
0 |
|
Dolor de cabeza |
18 |
2 |
|
|
Trastornos del metabolismo y la nutrición |
Disminución del apetito |
16 |
2 |
|
Neoplasias benignas, malignas, no especificadas |
Neoplasias secundarias* |
12* |
0 |
|
Trastornos vasculares |
Hipertensión |
16 |
8 |
|
*Un paciente murió debido a sarcoma histiocítico |
|||
Tabla 4: Disminución de hemoglobina, plaquetas o neutrófilos emergente* del tratamiento en pacientes con LLC/LLCP (N=51) en el Estudio 1
|
|
Porcentaje de pacientes (N = 51) |
|
|
|
Todos lo grados (%) |
Grado 3 o 4 (%) |
|
Disminución de plaquetas |
69 |
12 |
|
Disminución de neutrófilos |
53 |
26 |
|
Disminución de hemoglobina |
43 |
0 |
|
*Basado en las medidas de laboratorio de acuerdo con el criterio del Taller Internacional para LLC y las reacciones adversas. |
||
Estudio 2: Las reacciones adversas y anormalidades de laboratorio que se describen a continuación en las Tablas 5 y 6 reflejan la exposición a IMBRUVICA® con una mediana de duración de 8.6 meses y la exposición a ofatumumab con una mediana de 5.3 meses en el Estudio 2 en pacientes con LLC/LLCP previamente tratados.
|
Tabla 5: Reacciones adversas reportadas en ≥ 10% de los pacientes y al menos 2% mayor en el grupo tratado con IMBRUVICA® en pacientes con LLC/LLCP en el Estudio 2 |
||||
|
IMBRUVICA (N=195) |
Ofatumumab (N=191) |
|||
|
Sistema del cuerpo Reacción Adversa |
Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
48 |
4 |
18 |
2 |
|
Náuseas |
26 |
2 |
18 |
0 |
|
Estomatitis* |
17 |
1 |
6 |
1 |
|
Estreñimiento |
15 |
0 |
9 |
0 |
|
Vómitos |
14 |
0 |
6 |
1 |
|
Trastornos generales y condiciones en el lugar de la administración |
||||
|
Pirexia |
24 |
2 |
15 |
1 |
|
Infecciones e infestaciones |
||||
|
Infección del tracto respiratorio superior |
16 |
1 |
11 |
2 |
|
Neumonía* |
15 |
10 |
13 |
9 |
|
Sinusitis* |
11 |
1 |
6 |
0 |
|
Infección del tracto urinario |
10 |
4 |
5 |
1 |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Erupción* |
24 |
3 |
13 |
0 |
|
Petequias |
14 |
0 |
1 |
0 |
|
Hematomas* |
12 |
0 |
1 |
0 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||||
|
Dolor musculoesquelético* |
28 |
2 |
1 |
|
|
Artralgia |
17 |
1 |
0 |
|
|
Trastornos del sistema nervioso |
||||
|
Dolor de cabeza |
14 |
1 |
0 |
|
|
Mareos |
11 |
0 |
0 |
|
|
Lesiones, intoxicaciones y complicaciones del procedimiento |
||||
|
Contusión |
11 |
0 |
0 |
|
|
Trastornos oculares |
||||
|
Visión borrosa |
10 |
0 |
0 |
|
|
Los sujetos con múltiples eventos de un término de reacción adversa dado son contados una sola vez para cada término de la reacción adversa. Los términos de reacciones adversas individuales y del sistema del cuerpo se clasifican en orden descendente de frecuencia en el brazo de IMBRUVICA®. * Incluye múltiples términos de las reacciones adversas |
||||
|
Tabla 6: Disminución de hemoglobina, plaquetas o neutrófilos emergente* del tratamiento en pacientes con LLC/LLCP en el Estudio 2 |
||||
|
IMBRUVICA (n=195) |
Ofatumumab (N=191) |
|||
|
Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
|
Disminución de neutrófilos |
51 |
23 |
57 |
26 |
|
Disminución de plaquetas |
52 |
5 |
45 |
10 |
|
Disminución de hemoglobina |
36 |
0 |
21 |
0 |
|
* Basado en las mediciones de laboratorio según el criterio del Taller Internacional para la LLC. |
||||
Estudio 3: Las reacciones adversas descritas a continuación en la tabla 7 reflejan la exposición a IMBRUVICA® con una mediana de duración de 17.4 meses. La mediana de la exposición a clorambucil fue 7.1 meses en el Estudio 3.
|
Tabla 7: Reacciones adversas reportadas en ≥ 10% de los pacientes y al menos 2% mayor en el grupo tratado con IMBRUVICA® en pacientes con LLC/LLCP en el Estudio 3 |
||||
|
IMBRUVICA (N=135) |
Clorambucil (N=132) |
|||
|
Sistema del cuerpo Reacción Adversa |
Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
42 |
4 |
17 |
0 |
|
Estomatitis* |
14 |
1 |
4 |
1 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||||
|
Dolor musculoesquelético* |
36 |
4 |
20 |
0 |
|
Artralgia |
16 |
1 |
7 |
1 |
|
Espasmos musculares |
11 |
0 |
5 |
0 |
|
Trastornos oculares |
||||
|
Ojo seco |
17 |
0 |
5 |
0 |
|
Disminución del lagrimeo |
13 |
0 |
6 |
0 |
|
Visión borrosa |
13 |
0 |
8 |
0 |
|
Reducción de la agudeza visual |
11 |
0 |
2 |
0 |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Erupción* |
21 |
4 |
12 |
2 |
|
Hematomas* |
|
|
|
|
|
Infecciones e Infestaciones |
||||
|
Infección de la piel* |
15 |
2 |
3 |
1 |
|
Neumonía* |
14 |
8 |
7 |
4 |
|
Infecciones del tracto urinario |
10 |
1 |
8 |
1 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Tos |
22 |
0 |
15 |
0 |
|
Trastornos generales y condiciones e n el lugar de la administración |
||||
|
Edema periférico |
19 |
1 |
9 |
0 |
|
Pirexia |
17 |
0 |
14 |
2 |
|
Trastornos vasculares |
||||
|
Hipertensión* |
14 |
4 |
1 |
0 |
|
Trastornos del sistema nervioso |
||||
|
Dolor de cabeza |
12 |
1 |
10 |
2 |
|
Los sujetos con múltiples eventos de un término de reacción adversa dado son contados una sola vez para cada término de reacción adversa. Los términos de reacciones adversas individuales y del sistema del cuerpo se clasifican en orden descendente de frecuencia en el brazo de IMBRUVICA®. * Incluye múltiples términos de reacciones adversas |
||||
Estudio 4: Las reacciones adversas descritas a continuación en la tabla 8 reflejan la exposición a IMBRUVICA® + BR con una mediana de duración de 14.7 meses y exposición a placebo + BR con una mediana de 12.8 meses en el Estudio 4 en pacientes con LLC/LLCP previamente tratados.
|
Tabla 8: Reacciones adversas reportadas en al menos 10% de los pacientes y al menos 2% mayor en el grupo de IMBRUVICA® en pacientes con LLC/LLCP en el Estudio 4 |
||||
|
Ibrutinib + BR (N=287) |
Placebo + BR (N=287) |
|||
|
Sistema del cuerpo Reacción Adversa |
Todos los grados (%) |
Grado 3 4 (%) |
Todos los grados (%) |
Grado 3 4 (%) |
|
Trastornos del sistema linfático y sanguíneo |
||||
|
Neutropenia* |
66 |
61 |
60 |
55 |
|
Trombocitopenia* |
34 |
16 |
26 |
16 |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Erupción* |
32 |
4 |
25 |
1 |
|
Hematoma* |
20 |
< 1 |
8 |
< 1 |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
36 |
2 |
23 |
1 |
|
Dolor abdominal |
12 |
1 |
8 |
< 1 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||||
|
Dolor musculoesquelético* |
29 |
2 |
20 |
0 |
|
Espasmos musculares |
12 |
< 1 |
5 |
0 |
|
Trastornos generales y condiciones en el lugar de la administración |
||||
|
Pirexia |
25 |
4 |
22 |
2 |
|
Trastornos vasculares |
||||
|
Hemorragia* |
19 |
2 |
9 |
1 |
|
Hipertensión* |
11 |
5 |
5 |
2 |
|
Infecciones e infestaciones |
||||
|
Bronquitis |
13 |
2 |
10 |
3 |
|
Infección de la piel* |
10 |
3 |
6 |
2 |
|
Trastornos del metabolismo y la nutrición |
||||
|
Hiperuricemia |
10 |
2 |
6 |
0 |
|
Los términos de reacciones adversas individuales y del sistema del cuerpo se clasifican en orden descendente de frecuencia en el grupo de IMBRUVICA®. * Incluye múltiples términos de reacciones adversas < 1 usado para frecuencias mayores que 0 y menores que 0.5% |
||||
La fibrilación auricular de cualquier grado ocurrió en el 7% de los pacientes tratados con IMBRUVICA® + BR y en el 2% de los pacientes tratados con placebo + BR. La frecuencia de la fibrilación auricular de grado 3 y 4 fue 3% en los pacientes tratados con IMBRUVICA® + BR y 1% en los pacientes tratados con placebo + BR.
Macroglobulinemia de Waldenström y Linfoma de zona marginal: Los datos descritos a continuación reflejan la exposición a IMBRUVICA® en estudios clínicos abiertos que incluyeron 63 pacientes con MW previamente tratados (Estudio 5) y 63 pacientes con LZM previamente tratados (Estudio 6).
Las reacciones adversas más comunes que ocurrieron en los Estudios 5 y 6 (≥ 20%) fueron trombocitopenia, diarrea, neutropenia, fatiga, hematomas, hemorragias, anemia, erupción, dolor musculoesquelético y náuseas.
El nueve por ciento de los pacientes que recibieron IMBRUVICA® en los Estudios 5 y 6 discontinuaron el tratamiento debido a las reacciones adversas. Las reacciones adversas más comunes que condujeron a la discontinuación fueron enfermedad pulmonar intersticial, diarrea y erupción. Las reacciones adversas que condujeron a la reducción de la dosis ocurrieron en el 10% de los pacientes.
Estudio 5
Las reacciones adversas y anormalidades de laboratorio descritas a continuación en las tablas 9 y 10 reflejan la exposición a IMBRUVICA® con una mediana de duración de 11.7 meses en el Estudio 5.
|
Tabla 9: Reacciones adversas no hematológicas en ≥ 10% en pacientes con MW en el estudio 5 (N=63) |
|||
|
Sistema del cuerpo |
Reacción adversa |
Todos los grados (%) |
Grados 3 o 4 (%) |
|
Trastornos gastrointestinales |
Diarrea |
37 |
0 |
|
Náuseas |
21 |
0 |
|
|
Estomatitis* |
16 |
0 |
|
|
Enfermedad de reflujo gastroesofágico |
13 |
0 |
|
|
Trastornos de la piel y del tejido subcutáneo |
Erupción* |
22 |
0 |
|
Hematoma* |
16 |
0 |
|
|
Prurito |
11 |
0 |
|
|
Trastornos generales y condiciones en el lugar de la administración |
Fatiga |
21 |
0 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
Espasmos musculares |
21 |
0 |
|
Artropatía |
13 |
0 |
|
|
Trastornos respiratorios, torácicos y mediastínicos |
Tos |
19 |
0 |
|
Dolor orofaríngeo |
15 |
0 |
|
|
Disnea |
10 |
0 |
|
|
Infecciones e infestaciones |
Infección del tracto respiratorio superior |
19 |
0 |
|
Sinusitis |
19 |
0 |
|
|
Neumonía* |
14 |
6 |
|
|
Infecciones de la piel* |
14 |
2 |
|
|
Trastornos respiratorios, torácicos y mediastínicos |
Epistaxis |
19 |
0 |
|
Tos |
13 |
0 |
|
|
Trastornos del sistema nervioso |
Mareos |
14 |
0 |
|
Dolor de cabeza |
13 |
0 |
|
|
Neoplasias benignas, malignas y no especificados (incluyendo quistes y pólipos) |
Cáncer de piel* |
11 |
0 |
|
Los términos preferidos de las reacciones adversas individuales y del sistema del cuerpo se clasifican en orden descendente de frecuencia. * Incluye múltiples términos de reacciones adversas |
|||
Tabla 10: Disminución de hemoglobina, plaquetas o neutrófilos emergente* del tratamiento en pacientes con MW en el
Estudio 5 (N=63)
|
|
Porcentaje de pacientes (N = 63) |
|
|
Todos lo grados (%) |
Grado 3 o 4 (%) |
|
|
Disminución de plaquetas |
43 |
13 |
|
Disminución de neutrófilos |
44 |
19 |
|
Disminución de hemoglobina |
13 |
8 |
|
*Basado en las mediciones de laboratorio. |
||
Estudio 6: Las reacciones adversas y anormalidades de laboratorio descritas a continuación en las tablas 11 y 12 reflejan la exposición a IMBRUVICA® con una mediana de duración de 11.6 meses en el Estudio 6.
|
Tabla 11: Reacciones adversas no hematológicas en ≥ 10% de los pacientes con LZM en el Estudio 6 (N=63) |
|||
|
Sistema del cuerpo |
Reacción Adversa |
Todos los grados (%) |
Grados 3 o 4 (%) |
|
Trastornos gastrointestinales |
Diarrea |
43 |
5 |
|
Náuseas |
25 |
0 |
|
|
Dispepsia |
19 |
0 |
|
|
Estomatitis* |
17 |
2 |
|
|
Dolor abdominal |
16 |
2 |
|
|
Estreñimiento |
14 |
0 |
|
|
Dolor abdominal superior |
13 |
0 |
|
|
Vómito |
11 |
2 |
|
|
Trastornos generales y condiciones en el lugar de la administración |
Fatiga |
44 |
6 |
|
Edema periférico |
24 |
2 |
|
|
Pirexia |
17 |
2 |
|
|
Trastornos de la piel y del tejido subcutáneo |
Hematoma* |
41 |
0 |
|
Erupción* |
29 |
5 |
|
|
Prurito |
14 |
0 |
|
|
Trastornos musculoesqueléticos y del tejido conectivo |
Dolor musculoesquelético* |
40 |
3 |
|
Artralgia |
24 |
2 |
|
|
Espasmos musculares |
19 |
3 |
|
|
Infecciones e infestaciones |
Infección del tracto respiratorio superior |
21 |
0 |
|
Sinusitis* |
19 |
0 |
|
|
Bronquitis* |
11 |
0 |
|
|
Neumonía* |
11 |
10 |
|
|
Trastornos de la nutrición y del metabolismo |
Disminución del apetito |
16 |
2 |
|
Hiperuricemia |
16 |
0 |
|
|
Hipoalbuminemia |
14 |
0 |
|
|
Hipopotasemia |
13 |
0 |
|
|
Trastornos vasculares |
Hemorragia* |
30 |
0 |
|
Hipertensión |
14 |
5 |
|
|
Trastornos respiratorios, torácicos y mediastínicos |
Tos |
22 |
2 |
|
Disnea |
21 |
2 |
|
|
Trastornos del sistema nervioso |
Mareos |
19 |
0 |
|
Dolor de cabeza |
13 |
0 |
|
|
Trastornos psiquiátricos |
Ansiedad |
16 |
2 |
|
Los términos de reacciones adversas individuales preferidos y del sistema del cuerpo se clasifican en orden descendente de frecuencia. * Incluye múltiples términos de reacciones adversas |
|||
Tabla 12: Disminución de hemoglobina, plaquetas o neutrófilos emergente* del tratamiento en pacientes con LZM en el estudio 6 (N=63)
|
|
Porcentaje de pacientes (N = 63) |
|
|
Todos lo grados (%) |
Grado 3 o 4 (%) |
|
|
Disminución de plaquetas |
49 |
6 |
|
Disminución de hemoglobina |
43 |
13 |
|
Disminución de neutrófilos |
22 |
13 |
|
*Basado en las mediciones de laboratorio. |
||
Reacciones adversas adicionales importantes:
Diarrea: Ocurrió diarrea de cualquier grado a una tasa del 43% (rango, 36% a 59%) de los pacientes tratados con IMBRUVICA®. Ocurrió diarrea de grado 2 en el 9% (rango, 3% a 14%) y de grado 3 en el 3% (rango, 0 a 5%) de los pacientes tratados con IMBRUVICA®. La mediana del tiempo hasta la primera aparición de la diarrea de cualquier grado fue 10 días (rango, 0 a 627), de grado 2 fue 39 días (rango, 1 a 719) y de grado 3 fue 74 días (rango, 3 a 627). De los pacientes que reportaron diarrea, el 82% tuvo resolución completa, el 1% tuvo mejoría parcial y el 17% no reportó mejoría en el momento del análisis. La mediana del tiempo desde la aparición a la resolución o mejoramiento de la diarrea de cualquier grado fue 5 días (rango, 1 a 418) y fue similar para el grado 2 y 3. Menos del 1% de los pacientes discontinuaron el tratamiento con IMBRUVICA® debido a la diarrea.
Trastornos visuales: Visión borrosa y disminución de la agudeza visual de cualquier grado ocurrió en el 10% de los pacientes tratados con IMBRUVICA® (9% de grado 1, 2% de grado 2%). La mediana del tiempo hasta la primera aparición fue 85 días (rango, 1 a 414 días). De los pacientes con trastorno visual, el 61% tuvo resolución completa y el 38% no reportó mejoría en el momento del análisis. La mediana del tiempo desde la aparición hasta la resolución o el mejoramiento fue 29 días (rango, 1 a 335 días).
Experiencia posterior a la comercialización: Las siguientes reacciones adversas han sido identificadas durante el uso posterior a la aprobación de IMBRUVICA®. Debido a que estas reacciones son reportadas de forma voluntaria a partir de una población de tamaño no determinado, no siempre es posible estimar de forma confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos hepatobiliares: insuficiencia hepática: Trastornos respiratorios: enfermedad pulmonar intersticial
Trastornos metabólicos y de la nutrición: síndrome de lisis tumoral (ver sección Advertencias y precauciones)
Trastornos del sistema inmune: Shock anafiláctico, angioedema, urticaria.
Trastornos de la piel y del tejido subcutáneo: Síndrome de Stevens-Johnson, onicoclasis.
INTERACCIONES CON MEDICAMENTOS:
Inhibidores del CYP3A: Ibrutinib es metabolizado principalmente por la enzima 3A del citocromo P450 (CYP3A). En voluntarios sanos, la co-administración de ketoconazol, un inhibidor fuerte del CYP3A, incrementó la Cmáx y el AUC de ibrutinib en 29 y 24 veces, respectivamente. La dosis más alta de ibrutinib evaluada en los estudios clínicos fue 12.5 mg/kg (dosis actual de 840 – 1400 mg) administrada por 28 días con valores del AUC de dosis única de 1445 ± 869 ng.h/mL el cual es aproximadamente 50% mayor que las exposiciones en estado estacionario observadas con la dosis más alta indicada (560 mg).
Evitar la administración concomitante de IMBRUVICA® con inhibidores fuertes o moderados del CYP3A. Para inhibidores fuertes del CYP3A usados a corto plazo (por ejemplo antifúngicos y antibióticos por 7 días o menos, por ejemplo ketoconazol, itraconazol, voriconazol, posaconazol, claritromicina, telitromicina) considerar interrumpir la terapia con IMBRUVICA® durante la duración del uso del inhibidor.
Evitar los inhibidores fuertes del CYP3A que son crónicamente necesarios. Si un inhibidor moderado del CYP3A debe ser usado, reducir la dosis de IMBRUVICA®. Los pacientes que toman de forma concomitante inhibidores fuertes o moderados del CYP3A deben ser cercanamente monitoreados por signos de toxicidad con IMBRUVICA® (ver sección Posología y administración).
Evitar el pomelo y las naranjas de Sevilla durante el tratamiento con IMBRUVICA®, ya que estos contienen inhibidores moderados del CYP3A (ver sección Posología y administración y Farmacología clínica).
Inductores del CYP3A: La administración de IMBRUVICA® con rifampicina, un inductor fuerte del CYP3A, disminuyó la Cmáx y el AUC de ibrutinib aproximadamente en 13 y 10 veces respectivamente.
Evitar el uso concomitante de inductores fuertes del CYP3A (por ejemplo carbamezapina, rifampina, fenitoína y hierba de San Juan). Considerar agentes alternativos con menor inducción del CYP3A (ver sección Farmacología clínica).
ESTUDIOS CLÍNICOS:
Linfoma de células del manto: La seguridad y la eficacia de IMBRUVICA® en pacientes con LCM que han recibido al menos una terapia previa fueron evaluadas en un estudio abierto, multicéntrico, de un solo brazo de 111 pacientes previamente tratados. La mediana de la edad fue 68 años (rango, 40 a 84 años); el 77% fueron varones y el 92% fueron Caucásicos. En condiciones basales, el 89% de los pacientes tuvo un estado de desempeño basal en la escala ECOG de 0 o 1. La mediana del tiempo desde el diagnóstico fue 42 meses, y la mediana del número de tratamientos previos fue 3 (rango, 1 a 5 tratamientos), incluyendo 11% con trasplante previo de célula madre. En condiciones basales, el 39% de los sujetos tuvo al menos un tumor ≥ 5 cm, el 49% tuvo compromiso de la médula ósea, y el 54% tuvo compromiso extranodal en la exploración.
IMBRUVICA se administró por vía oral en dosis de 560 mg una vez al día hasta la progresión de la enfermedad o una toxicidad inaceptable. La respuesta tumoral se evaluó según los criterios revisados por el Grupo Internacional de Trabajo (IWG) para el linfoma no-Hodgkin (LNH). El criterio principal de valoración en este estudio fue la tasa de respuesta global (TRG) evaluado por el investigador. Las respuestas a IMBRUVICA® se muestran en la Tabla 13.
Tabla 13: Tasa de respuesta global (TRG) y duración de la respuesta (DR) basados en la evaluación del investigador en pacientes con LCM
|
|
Total (N=111) |
|
TRG (%) |
65.8 |
|
IC al 95% (%) |
(56.2; 74.5) |
|
RC (%) |
17.1 |
|
RP (%) |
48.6 |
|
Mediana de la DR meses (IC al 95%) |
17.5 (15.8; NA) |
|
IC = Intervalo de confianza; RC = Respuesta completa; RP = Respuesta parcial; NA = No alcanzada |
|
Un Comité de Revisión Independiente (CRI) realizó la lectura e interpretación independiente de las imágenes. La revisión del CRI demostró un TRG del 69%.
La mediana del tiempo de respuesta fue 1.9 meses.
Linfocitosis: Al inicio con IMBRUVICA®, ocurrió un incremento temporal en el recuento de linfocitos (es decir, incremento ≥ 50% desde el valor basal y por encima del recuento absoluto de linfocitos de 5 000/mcL) en el 33% de los pacientes en el estudio de LCM. La aparición de linfocitosis aislada ocurre durante las primeras semanas de terapia con IMBRUVICA® y se resuelve en una mediana de 8 semanas.
Leucemia linfocítica crónica/Linfoma linfocítico de células pequeñas: La seguridad y eficacia de IMBRUVICA® en pacientes con LLC/LLCP fueron demostradas en un estudio no controlado y tres estudios aleatorizados controlados.
Estudio 1: Se realizó un estudio abierto, multicéntrico en 48 pacientes con LLC previamente tratados. La mediana de la edad fue 67 años (rango, 37 a 82 años), el 71% fueron varones y el 94% fueron Caucásicos. Todos los pacientes tuvieron un estado de desempeño basal del ECOG de 0 o 1. La mediana del tiempo desde el diagnóstico fue de 80 meses y la mediana del número de tratamientos previos fue 4 (rango, 1 a 12 tratamientos). En condiciones basales, el 46 % de los sujetos tuvo al menos un tumor ≥ 5 cm.
IMBRUVICA® se administró por vía oral a 420 mg una vez al día hasta la progresión de la enfermedad o toxicidad inaceptable. El TRG y la DR fueron evaluados usando una versión modificada del Taller Internacional sobre el criterio para el LLC por un Comité de Revisión Independiente. El TRG fue 58.3% (IC al 95%: 43.2%, 72.4%), todas respuestas parciales. Ninguno de los pacientes alcanzó una respuesta completa. La DR varió de 5.6 a 24.2+ meses. No se alcanzó la mediana de la DR.
Estudio 2: Se realizó un estudio abierto, multicéntrico, aleatorizado, Fase 3, de IMBRUVICA® en comparación con ofatumumab en pacientes con LLC o LLCP previamente tratados. Los pacientes (n=391) fueron aleatorizados 1:1 para recibir 420 mg de IMBRUVICA® una vez al día hasta la progresión de la enfermedad o toxicidad inaceptable, u ofatumumab a una dosis inicial de 300 mg, seguido una semana después por una dosis semanal de 2000 mg por 7 dosis y luego cada 4 semanas por 4 dosis adicionales. Cincuenta y siete pacientes aleatorizados a ofatumumab fueron cambiados para recibir IMBRUVICA® después de la progresión. La mediana de la edad fue 67 años (rango, 30 a 88 años), el 68% fueron varones y el 90% fueron Caucásicos. Todos los pacientes tuvieron un estado de desempeño basal del ECOG de 0 o 1. El estudio enroló 373 pacientes con LLC y 18 pacientes con LLCP. La mediana del tiempo desde el diagnóstico fue 91 meses y la mediana del número de tratamientos previos fue 2 (rango, 1 a 13 tratamientos). En condiciones basales, el 58 % de los pacientes tuvo al menos un tumor ≥ 5 cm. El treinta y dos por ciento de los pacientes tuvieron deleción del 17p.
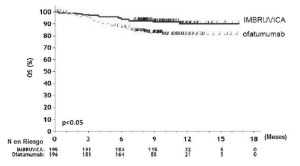
Los resultados de la eficacia del Estudio 2 se muestran en la tabla 14 y las curvas de KaplanMeier de la supervivencia libre de progresión (SLP) evaluada por un CRI de acuerdo al criterio del Taller Internacional para LLC y la supervivencia global (SG) se muestran en las figuras 1 y 2, respectivamente.
Tabla 14: Resultados de eficacia en pacientes con LLC/LLCP en el estudio 2
|
Criterio de valoración |
IMBRUVICA N=195 |
Ofatumumab N=196 |
|
Supervivencia libre de progresiónb |
||
|
Número de eventos (%) |
35 (17.9) |
111 (56.6) |
|
Progresión de la enfermedad |
26 |
93 |
|
Eventos de muerte |
9 |
18 |
|
Mediana (IC al 95%), meses |
NA |
8.1 (7.2, 8.3) |
|
HR (IC al 95%) |
0.22 (0. 15, 0.32) |
|
|
Supervivencia globala |
||
|
Número de muertes (%) |
16 (8.2) |
33 (16.8) |
|
HR (IC al 95%) |
0.43 (0.24, 0.79) |
|
|
Tasa de Respuesta Globalb |
42.6% |
4.1% |
|
a Mediana de la supervivencia global no alcanzada en ningún grupo b Evaluada por el CRI. Se alcanzaron todas las respuestas parciales; ninguno de los pacientes alcanzó una respuesta completa. IC: Intervalo de confianza; HR = cociente de riesgo; NA = No alcanzada. |
||
Figura 1: Curva de Kaplan-Meier de supervivencia libre de progresión [población con intención de tratar] en pacientes con LLC/LLCP en el Estudio 2
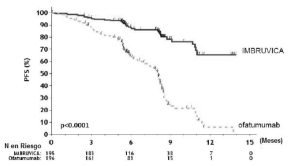
Figura 2: Curva de Kaplan-Meier de supervivencia global (población con intención de tratar) en pacientes con LLC/LLCP en el Estudio 2
LLC/LLCP con deleción del 17p (LLC/LLCP con del 17p) en el Estudio 2: El estudio 2 incluyó 127 pacientes con LLC/LLCP con deleción del 17p. La mediana de la edad fue 67 años (rango, 30 a 84 años), el 62% fueron varones y el 88% fueron Caucásicos. Todos los pacientes tuvieron un estado de desempeño basal del ECOG de 0 o 1. La SLP y la TRG fueron evaluados por el CRI. Los resultados de la eficacia para el LLC/LLCP con deleción del 17p se muestran en la Tabla 15.
Tabla 15: Resultados de la eficacia en pacientes con LLC/LLC con deleción del 17p en el Estudio 2
|
Criterio de valoración |
IMBRUVICA N=63 |
Ofatumumb N=64 |
|
Supervivencia libre de progresióna |
||
|
Número de eventos (%) |
16 (25.4) |
38 (59.4) |
|
Progresión de la enfermedad |
12 |
31 |
|
Eventos de muerte |
4 |
7 |
|
Mediana (IC al 95%), meses |
NA |
|
|
HR (IC al 95%) |
0.25 (0. 14, 0.45) |
|
|
Tasa de Respuesta Globala |
47.6% |
4.7% |
|
a Evaluada por el CRI. Todas las respuestas alcanzadas fueron parciales; ninguno de los pacientes alcanzó una respuesta completa. IC: Intervalo de confianza; HR = cociente de riesgo; NA = No alcanzada. |
||
Estudio 3: Se condujo un estudio abierto, aleatorizado, multicéntrico de IMBRUVICA® en comparación con clorambucil en pacientes con LLC o LLCP sin tratamiento previo de 65 años de edad o mayores. Los pacientes (n = 269) fueron aleatorizados 1:1 para recibir 420 mg de IMBRUVICA® al día hasta la progresión de la enfermedad o toxicidad inaceptable, o clorambucil a una dosis inicial de 0.5 mg/kg en los días 1 y 15 de cada ciclo de 28 días durante un máximo de 12 ciclos, con un margen de incremento de dosis intrapaciente hasta 0.8 mg/kg en base a la tolerabilidad.
La mediana de la edad fue 73 años (rango, 65 a 90 años), el 63% fueron varones y el 91% fueron Caucásicos.
El noventa y uno por ciento de los pacientes tuvo un estado de desempeño basal del ECOG de 0 o 1 y el 9% tuvo un estado de desempeño basal del ECOG de 2. El ensayo enroló a 249 pacientes con LLC y 20 pacientes con LLCP. En condiciones basales, el 20% de los pacientes tuvo deleción del 11q. Las razones más comunes para iniciar la terapia de la LLC incluyen: insuficiencia medular progresiva demostrada por la anemia y/o trombocitopenia (38%), linfadenopatía progresiva o sintomática (37%), esplenomegalia progresiva o sintomática (30%), fatiga (27%) y sudoración nocturna (25%).
Con una mediana del seguimiento de 28.1 meses, hubo 32 eventos de muerte observados [11 (8.1%) y 21 (15.8%) en los brazos de tratamiento de IMBRUVICA® y clorambucil, respectivamente]. Con el 41% de los pacientes que cambiaron de clorambucil a IMBRUVICA el análisis de supervivencia global en la población de pacientes con intención de tratar produjo un HR estadísticamente significativo de 0.44 [IC al 95% (0.21, 0.92)] y estimados de la tasa de supervivencia a 2 años de 94.7% [IC al 95% (89.1, 97.4)] y 84.3% [IC al 95% (76.7, 89.6)] en los grupos de IMBRUVICA® y clorambucil, respectivamente.
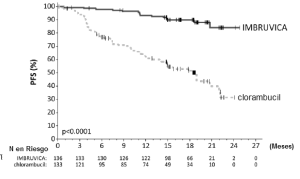
Los resultados de la eficacia del Estudio 3 se muestran en la tabla 16 y las curvas de KaplanMeier para la SLP evaluada por un CRI de acuerdo con el criterio del Taller Internacional para LLC se muestran en la Figura 3.
Tabla 16: Resultados de eficacia en pacientes con LLC/LLCP en el Estudio 3
|
Criterio de valoración |
IMBRUVICA® N=136 |
Clorambucil N=133 |
|
Supervivencia libre de progresióna |
||
|
Número de eventos (%) |
15 (11.0) |
64 (48.1) |
|
Progresión de la enfermedad |
12 |
57 |
|
Eventos de muerte |
3 |
7 |
|
Mediana (IC al 95%), meses |
NA |
18.9 (14.1, 22.0) |
|
HRb (IC al 95%) |
0.1 6 (0.09, 0.28) |
|
|
Tasa de Respuesta Globala (RC + RP) |
82.4% |
35.3% |
|
Valor – p |
< 0.0001 |
|
|
a Evaluada por el CRI: 5 sujetos (3.7%) en el brazo de IMBRUVICA ® y dos sujetos (1.5%) en el grupo de clorambucil alcanzaron respuesta completa b HR = cociente de riesgo; NA = No alcanzada. |
||
Figura 3: Curva de Kaplan-Meier de supervivencia libre de progresión (población con intención de tratar) en pacientes con LLC/LLCP en el Estudio 2
Estudio 4: Se realizó un estudio aleatorizado, multicéntrico, doble ciego, Fase 3 de IMBRUVICA® en combinación con bendamustina y rituximab (BR) en comparación con el placebo + BR en pacientes con LLC o LLCP previamente tratados. Los pacientes (n=578) fueron aleatorizados 1:1 para recibir 420 mg de IMBRUVICA® al día o el placebo en combinación con BR hasta la progresión de la enfermedad o toxicidad inaceptable. Todos los pacientes recibieron BR por un máximo de seis ciclos de 28 días. Bendamustina fue dosificada a 70 mg/m2 en infusión IV en 30 minutos en el ciclo 1, los días 2 y 3 y en los ciclos 2-6, los días 1 y 2 hasta por 6 ciclos. Rituximab se administró a una dosis de 375 mg/m2 en el primer ciclo, el día 1, y 500 mg/m2 en el ciclo 2 hasta el 6, el día 1.
La mediana de la edad fue 64 años (rango, 31 a 86 años), el 66% fueron hombres y el 91% Caucásicos. Todos los pacientes tuvieron un estado de desempeño basal del ECOG de 0 o 1. La mediana de tiempo desde el diagnóstico fue 5.9 años y la mediana del número de tratamientos previos fue 2 (rango, 1 a 11 tratamientos). En condiciones basales, el 56% de los pacientes tuvo al menos un tumor ≥ 5 cm y el 26% presentaron deleción del 11q.
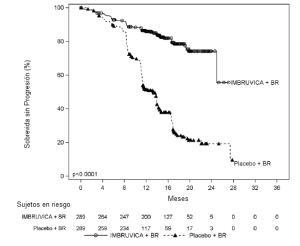
Los resultados de la eficacia para el Estudio 4 se muestran en la tabla 17 y las curvas de Kaplan-Meier para SLP se muestran en la Figura 4.
Tabla 17: Resultados de eficacia en pacientes con LLC/LLCP en el Estudio 4
|
Criterio de valoración |
IMBRUVICA + BR N=289 |
Placebo + BR N=289 |
|
Supervivencia libre de progresióna |
||
|
Número de eventos (%) |
56 (19.4) |
183 (63.3) |
|
Mediana (IC al 95%), meses |
No se alcanzó |
13.3 (11.3, 13.9) |
|
HR (IC al 95%) |
0.20 (0. 15, 0.28) |
|
|
Tasa de respuesta globala |
82.7% |
67.8% |
|
a Evaluada por el CRI, veinticuatro sujetos (8.3%) en el brazo de IMBRUVICA® + BR y seis sujetos (2.1%) en el brazo del placebo + BR alcanzaron respuesta completa. BR= bendamustina y rituximab; IC = intervalo de confianza, HR= cociente de riesgo. |
||
Figura 4: Curva de Kaplan-Meier de supervivencia libre de progresión (Población con intención de tratar) en pacientes con LLC/LLCP en el Estudio 4
Linfocitosis: Después del inicio con IMBRUVICA®, ocurrió un incremento en el recuento de linfocitos (es decir, incremento ≥ 50% desde las condiciones basales y por encima del recuento absoluto de linfocitos de 5000/mcL) en el 66% de los pacientes en los estudios de LLC. La aparición de la linfocitosis aislada ocurre durante el primer mes de la terapia con IMBRUVICA® y se resuelve en una mediana de 14 semanas (rango, 0.1 a 104 semanas). Cuando IMBRUVICA® se administró con la quimioinmunoterapia, la linfocitosis fue 7% con IMBRUVICA® + BR en comparación con el 6% con placebo + BR.
Macroglobulinemia de Waldenström: La seguridad y eficacia de IMBRUVICA® en la MW fueron evaluadas en un ensayo abierto, multicéntrico, de un solo grupo de 63 pacientes previamente tratados. La mediana de la edad fue 63 años (rango, 44 a 86 años), el 76% fueron varones y 95% fueron Caucásicos. Todos los pacientes tuvieron un estado de desempeño basal del ECOG de 0 o 1. La mediana del tiempo desde el diagnóstico fue 74 meses y la mediana del número de tratamientos previos fue 2 (rango, 1 a 11 tratamientos). En condiciones basales, la mediana del valor sérico de la IgM fue 3.5g/dL (rango, 0.7 a 8.4 g/dL).
IMBRUVICA® fue administrado por vía oral a 420 mg una vez al día hasta la progresión de la enfermedad o toxicidad inaceptable. Las respuestas fueron evaluadas por los investigadores y un CRI usando el criterio adoptado del Grupo de trabajo Internacional para la Macroglobulinemia de Waldenström. Las respuestas, definidas como respuestas parciales o mejores, por el CRI se muestran en la Tabla 18.
Tabla 18. Tasa de respuesta global (TRG) y duración de la respuesta (DR) basada en la evaluación del CRI en pacientes con MW en el Estudio 5
|
|
Total ( N= 63) |
|
Tasa de Respuesta (RC+PRMB+RP), % |
61.9 |
|
IC al 95% (%) |
(48.8, 73.9) |
|
Respuesta completa (CR) |
0 |
|
Respuesta parcial muy buena (PRMB) % |
11.1 |
|
Respuesta parcial (RP), (%) |
50.8 |
|
Mediana de la duración de la respuesta, meses (rango) |
NA (2.8 + , 18.8+) |
|
IC = Intervalo de confidencia; NA = No alcanzado |
|
La mediana del tiempo hasta la respuesta fue 1.2 meses (rango, 0.7 – 13.4 meses)
Linfoma de Zona Marginal: La seguridad y eficacia de IMBRUVICA® en el LZM fueron evaluadas en un estudio abierto, multicéntrico, de un grupo de pacientes que recibieron al menos una terapia previa. El análisis de la eficacia incluyó 63 pacientes con 3 sub-tipos de LZM: tejido linfoide asociado a la mucosa (MALT; N=32), nodal (N=17) y esplénico (N=14). La mediana de la edad fue de 66 años (rango, 30 a 92 años), el 59% fueron mujeres, y el 84% fueron Caucásicas. El noventa y dos por ciento de las pacientes tuvo un estado de desempeño basal del ECOG de 0 o 1y el 8% tuvo un estado de desempeño basal del ECOG de 2. La mediana del tiempo desde el diagnóstico fue de 3.8 años, y la mediana del número de tratamientos previos fue 2 (rango, 1 a 9 tratamientos).
IMBRUVICA® se administró vía oral a 560 mg una vez al día hasta la progresión de la enfermedad o toxicidad inaceptable. Las respuestas fueron evaluadas por los investigadores y un CRI usando el criterio adoptado del Grupo de Trabajo Internacional para el linfoma maligno. Las respuestas del CRI se muestran en la tabla 19.
Tabla 19. Tasa de respuesta global (TRG) y duración de la respuesta (DR) basada en la evaluación del CRI en pacientes con LZM en el Estudio 6
|
|
Total (N= 63) |
|
Tasa de respuesta (RC+RP), % |
46.0 % |
|
IC al 95%(%) |
(33.4, 59.1) |
|
Respuesta completa (CR) |
3.2 |
|
Respuesta parcial (PR) % |
42.9 |
|
Mediana de la duración de la respuesta, meses (rango) |
NR (16.7, NR) |
|
IC = Intervalo de confidencia; NA = No alcanzado |
|
Mediana del tiempo de seguimiento del estudio= 19.4 meses
La mediana del tiempo hasta la respuesta fue 4.5 meses (rango, 2.3 - 16.4 meses). Las tasas de respuesta global fueron 46.9%, 41.2% y 50.0% para los 3 sub-tipos de LZM (MALT, nodal y esplénico), respectivamente.
TOXICOLOGÍA NO CLÍNICA:
Carcinogénesis, mutagénesis, deterioro de la fertilidad: No se han realizado estudios de carcinogénesis con ibrutinib.
Ibrutinib no fue mutagénico en un ensayo de mutagenicidad bacteriana (Ames), no fue clastogénico en un ensayo de aberración cromosómica en células mamíferas (CHO), ni fue clastogénico en un ensayo in vivo de micronúcleos en médula ósea en ratones a dosis hasta de 2000 mg/kg.
Se administró a ratas dosis orales diarias de ibrutinib, por 4 semanas antes y durante el apareamiento en los machos y por 2 semanas antes y durante el apareamiento en las hembras. El tratamiento de las hembras continuó después del embarazo hasta el día 7 de la gestación (GD), y el tratamiento de los machos continuó hasta el final del estudio. No se observaron efectos sobre la fertilidad o capacidad reproductiva en ratas machos o hembras hasta la dosis máxima ensayada, 100 mg/kg/día (Dosis Humana Equivalente [DHE] 16 mg/kg).
ADVERTENCIAS Y PRECAUCIONES:
Hemorragia: Eventos hemorrágicos fatales ocurrieron en pacientes tratados con IMBRUVICA®. Eventos hemorrágicos de grado 3 o mayores (hemorragia intracraneal [incluyendo hematoma subdural], sangrado gastrointestinal, hematuria y hemorragia post procedimiento) ocurrieron hasta en el 6% de los pacientes. Los eventos hemorrágicos de cualquier grado, incluyendo hematomas y petequias, ocurrieron en aproximadamente la mitad de los pacientes tratados con IMBRUVICA®.
No se conoce bien el mecanismo de los eventos hemorrágicos.
IMBRUVICA® puede incrementar el riesgo de hemorragia en pacientes que reciben terapias con anticoagulantes o antiplaquetarios y los pacientes deben ser monitoreados por los signos de sangrado.
Considerar el riesgo-beneficio de suspender IMBRUVICA® por lo menos 3 a 7 días antes y después de la cirugía dependiendo del tipo de cirugía y del riesgo de sangrado (ver sección Estudios clínicos).
Infecciones: Infecciones fatales y no fatales han ocurrido con la terapia con IMBRUVICA®. Infecciones de grado 3 o mayores ocurrieron en el 14% al 29% de los pacientes (ver sección Reacciones adversas). Casos de leucoencefalopatía multifocal progresiva (LMP) y neumonía por Pneumocystis jirovecii (PJP) han ocurrido en pacientes tratados con IMBRUVICA®. Evaluar a los pacientes por fiebre e infecciones y tratar adecuadamente.
Citopenias: Citopenias de grado 3 o 4 emergentes del tratamiento incluyendo neutropenia (rango, 13 a 29%), trombocitopenia (rango, 5 a 17%) y anemia (rango, de 0 a 13%) basados en medidas de laboratorio ocurrieron en pacientes tratados con IMBRUVICA® en monoterapia.
Monitorear mensualmente los recuentos sanguíneos completos.
Fibrilación auricular: Fibrilación auricular y aleteo auricular (rango, de 6 a 9%) han ocurrido en pacientes tratados con IMBRUVICA®, especialmente en pacientes con factores de riesgo cardíaco, hipertensión, infecciones agudas y antecedentes previos de fibrilación auricular. Monitorear de forma periódica y clínica a los pacientes por fibrilación auricular. Los pacientes que desarrollan síntomas arrítmicos (por ejemplo, palpitaciones, aturdimiento) o la aparición de disnea deben someterse a un electrocardiograma (ECG). La fibrilación auricular debe ser manejada apropiadamente, y si persiste, considerar los riesgos y beneficios del tratamiento con IMBRUVICA® y seguir las guías de modificación de la dosis (ver sección Posología y administración).
Hipertensión: Ha ocurrido hipertensión (rango, 6 a 17%) en los pacientes tratados con IMBRUVICA® con una mediana del tiempo hasta la aparición de 4.6 meses (rango, 0.03 a 22 meses). Monitorear a los pacientes por aparición de hipertensión o hipertensión que no está adecuadamente controlada después de iniciar IMBRUVICA®.
Ajustar los medicamentos antihipertensivos existentes y/o iniciar tratamiento antihipertensivo según sea apropiado.
Segundas neoplasias primarias: Otras neoplasias (rango, 3 a 16%) incluyendo carcinomas no cutáneos (rango, 1 a 4%) han ocurrido en pacientes tratados con IMBRUVICA®. La segunda neoplasia primaria más frecuente fue el cáncer de piel no melanoma (rango, 2 a 13%).
Síndrome de lisis tumoral: Se ha reportado de manera poco frecuente síndrome de lisis tumoral con la terapia con IMBRUVICA®. Evaluar el riesgo en condiciones basales (por ejemplo, carga tumoral elevada) y tomar las precauciones adecuadas. Monitorear cercanamente a los pacientes y tratar según sea apropiado.
Toxicidad embrio-fetal: En base a los hallazgos en animales, IMBRUVICA® puede causar daños fetales cuando se administra a mujeres embarazadas. La administración de ibrutinib a conejos y ratas gestantes durante el período de organogénesis causó toxicidad embriofetal incluyendo malformaciones a exposiciones que fueron de 2-20 veces mayores que las reportadas en pacientes con neoplasias hematológicas. Advertir a las mujeres de evitar el embarazo mientras toman IMBRUVICA® y durante un mes después de la culminación de la terapia. Si este fármaco se usa durante el embarazo o si la paciente queda embarazada mientras está tomando este fármaco, la paciente debe ser informada del riesgo potencial para el feto (ver sección Uso en poblaciones específicas).
POSOLOGÍA Y ADMINISTRACIÓN:
Guías de dosificación: Administrar IMBRUVICA® por vía oral una vez al día aproximadamente a la misma hora cada día. Ingerir las cápsulas enteras con agua. No abrir, romper ni masticar las cápsulas.
Posología:
• Linfoma de células del manto y Linfoma de zona marginal: La dosis recomendada de IMBRUVICA® para el LCM y LZM es 560 mg (cuatro cápsulas de 140 mg) por vía oral una vez al día hasta la progresión de la enfermedad o toxicidad inaceptable.
• Leucemia linfocítica crónica/Linfoma linfocítico de células pequeñas y macroglobulinemia de Waldenström: La dosis recomendada de IMBRUVICA® para la LLC/LLCP y la MW es 420 mg (tres cápsulas de 140 mg) por vía oral una vez al día hasta la progresión de la enfermedad o toxicidad inaceptable.
La dosis recomendada de IMBRUVICA® para la LLC/LLCP cuando es usado en combinación con bendamustina y rituximab (administrado cada 28 días hasta por 6 ciclos) es 420 mg (tres cápsulas de 140 mg) por vía oral una vez al día hasta la progresión de la enfermedad o toxicidad inaceptable.
• Modificación de la dosis por reacciones adversas: Interrumpir el tratamiento con IMBRUVICA® por cualquier toxicidad no hematológica de grado 3 o mayor, neutropenia de grado 3 o mayor con infección o fiebre, o toxicidad hematológica de grado 4. Una vez que los síntomas de toxicidad se han resuelto hasta llegar al grado 1 o al valor basal (recuperación), el tratamiento con IMBRUVICA® se puede reiniciar con la dosis inicial. Si reaparece la toxicidad, reduzca la dosis en una cápsula (140 mg por día). Se puede considerar una segunda reducción de dosis en 140 mg si es necesario. Si estas toxicidades persisten o reaparecen luego de dos reducciones de dosis, discontinuar el tratamiento con IMBRUVICA®.
A continuación se describen las modificaciones de la dosis recomendada:
|
Ocurrencia de la toxicidad |
Modificación de la dosis para el LCM y LZM después de la recuperación Dosis inicial = 560 mg |
Modificación de la dosis para la LLC/LLCP y MW después de la recuperación Dosis inicial = 420 mg |
|
Primera |
Reiniciar a 560 mg diarios |
Reiniciar a 420 mg diarios |
|
Segunda |
Reiniciar a 420 mg diarios |
Reiniciar a 280 mg diarios |
|
Tercera |
Reiniciar a 280 mg diarios |
Reiniciar a 140 mg diarios |
|
Cuarta |
Discontinuar IMBRUVICA |
Discontinuar IMBRUVICA |
Modificación de la dosis para el uso con inhibidores del CYP3A: Evitar la co-administración con inhibidores fuertes o moderados del CYP3A y considerar agentes alternativos con menor inhibición del CYP3A.
No se recomienda el uso concomitante de inhibidores fuertes del CYP3A que deben ser tomados en forma crónica (por ejemplo, ritonavir, indinavir, nelfinavir, saquinavir, boceprevir, telaprevir, nefazodona). Para el uso a corto plazo (tratamiento por 7 días o menos) de inhibidores fuertes del CYP3A (por ejemplo, antifúngicos y antibióticos) considerar la interrupción de la terapia con IMBRUVICA® hasta que el inhibidor del CYP3A ya no sea necesario (ver sección Interacción con medicamentos).
Reducir la dosis de IMBRUVICA® hasta 140 mg si un inhibidor moderado del CYP3A debe ser usado (por ejemplo, fluconazol, darunavir, eritromicina, diltiazen, atazanavir, aprepitant, amprenavir, fosamprenavir, crizotinib, imatinib, verapamilo y ciprofloxacina) (ver sección Interacción con medicamentos).
Los pacientes que están tomando de forma concomitante inhibidores fuertes o moderados del CYP3A deben ser monitoreados más cercanamente por los signos de toxicidad con IMBRUVICA®.
Modificación de la dosis para el uso en insuficiencia hepática: Para pacientes con insuficiencia hepática leve (Child-Pugh, clase A), la dosis recomendada es 140 mg diarios (una cápsula). Evitar el uso de IMBRUVICA® en pacientes con insuficiencia hepática moderada o severa (Child-Pugh, clase B y C) (ver sección Uso en Poblaciones específicas y Farmacología clínica).
Dosis omitida: Si una dosis de IMBRUVICA® no se toma a la hora programada, puede ser tomada lo antes posible el mismo día retomando el horario normal al día siguiente. No se deben tomar cápsulas adicionales de IMBRUVICA® para compensar la dosis omitida.
USO EN POBLACIONES ESPECÍFICAS:
Embarazo:
Resumen de riesgo: IMBRUVICA®, un inhibidor de quinasa, puede causar daño fetal basado en los hallazgos de los estudios con animales. En los estudios de reproducción animal, la administración de ibrutinib a conejos y ratas gestantes durante el período de organogénesis a exposiciones hasta de 2-20 veces la dosis clínica de 420-560 mg al día produjo toxicidad embriofetal incluyendo malformaciones (ver sección Datos en animales). Si IMBRUVICA® es usado durante el embarazo o si la paciente queda embarazada mientras está tomando
IMBRUVICA®, la paciente debe ser advertida del riesgo potencial al feto.
Se desconoce el riesgo estimado de los antecedentes de los principales defectos del nacimiento y del aborto espontáneo de la población indicada. En la población general de los EE.UU., el riesgo estimado de los antecedentes de los principales defectos del nacimiento y del aborto espontáneo en los embarazos clínicamente reconocidos es de 2-4% y 15-20%, respectivamente.
Datos en animales: Ibrutinib fue administrado oralmente a ratas gestantes durante el período de organogénesis con dosis orales de 10, 40 y 80 mg/kg/día. Ibrutinib a dosis de 80 mg/kg/día fue asociado con malformaciones viscerales (corazón y los vasos principales) y con el incremento de la resorción y las pérdidas posteriores a la implantación. La dosis de 80 mg/kg/día en ratas es aproximadamente 14 veces la exposición (AUC) en paciente con LCM y 20 veces la exposición en pacientes con LLC/LLCP o MW que recibieron dosis de 560 mg y 420 mg una vez al día, respectivamente. Ibrutinib a dosis de 40mg/kg/día o mayores fueron asociados con la disminución del peso fetal. La dosis de 40 mg/kg/día en ratas es aproximadamente 6 veces la exposición (AUC) en pacientes con LCM que recibieron 560 mg una vez al día.
Ibrutinib también fue administrado oralmente a conejos gestantes durante el período de organogénesis a dosis de 5, 15 y 45 mg/kg/día. Ibrutinib a dosis de 15 mg/kg/día o mayores fue asociada con variaciones esqueléticas (esternebras fundidas) e ibrutinib a dosis de 45 mg/kg/día fue asociado con el incremento de la resorción y pérdidas posteriores a la implantación. La dosis de 15 mg/kg/día en conejos es aproximadamente 2.0 veces la exposición (AUC) en pacientes con LCM y 2.8 veces la exposición en pacientes con LLC/LLCP o MW que recibieron dosis de 560 y 420 mg al día, respectivamente.
Lactancia:
Resumen del riesgo: No existe información relacionada a la presencia de ibrutinib o sus metabolitos en la leche humana, los efectos en el lactante o sus efectos en la producción de la leche.
El desarrollo y los beneficios para la salud de la lactancia deben considerarse junto con la necesidad clínica de la madre de IMBRUVICA® y los posibles efectos adversos en el niño amamantado o de la condición materna subyacente.
Potencial reproductivo en varones y mujeres:
Prueba de embarazo: Verificar el estado de embarazo de las mujeres de potencial reproductivo antes de iniciar la terapia con IMBRUVICA®.
Anticoncepción:
Mujeres: Advertir a las mujeres con potencial reproductivo evitar el embarazo mientras toman IMBRUVICA® y hasta 1 mes después de la finalización del tratamiento. Si se utiliza este fármaco durante el embarazo o si la paciente queda embarazada mientras toma este fármaco, la paciente debe ser informada del riesgo potencial para un feto.
Varones: Advertir a los varones evitar la procreación mientras reciben IMBRUVICA®, y por 1 mes después de la última dosis de IMBRUVICA®.
Uso pediátrico: No se ha establecido la seguridad ni la eficacia de IMBRUVICA® en niños.
Uso geriátrico: De los 905 pacientes en los estudios clínicos con IMBRUVICA®, el 62% fueron ≥ 65 años de edad, mientras que el 21% fueron ≥ 75 años de edad. En general no se observaron diferencias en la eficacia entre pacientes jóvenes y pacientes mayores. Anemia (todos los grados) y neumonía de grado 3 o mayor ocurrió con mayor frecuencia entre los pacientes mayores tratados con IMBRUVICA®.
Insuficiencia hepática: Ibrutinib es metabolizado en el hígado. En un estudio de insuficiencia hepática, los datos mostraron un incremento en la exposición a ibrutinib. Después de la administración de una dosis única, el AUC de ibrutinib se incrementó en 2.7 – 8.2 y 9.8 veces en sujetos con insuficiencia hepática leve (Child-Pugh clase A), moderada (Child-Pugh clase B) y severa (Child-Pugh clase C) comparado con sujetos con función hepática normal.
La seguridad de IMBRUVICA® no ha sido evaluada en pacientes con cáncer con insuficiencia hepática leve a severa según los criterios de Child-Pugh.
Monitorear a los pacientes por signos de toxicidad con IMBRUVICA® y seguir la guía de modificación de dosis según sea necesario. No se recomienda administrar IMBRUVICA® a pacientes con insuficiencia hepática moderada o severa (Child-Pugh clase B y C) (ver sección Posología y Administración y Farmacología clínica).
Plasmaféresis: El manejo de la hiperviscosidad en pacientes con MW puede incluir plasmaféresis antes y durante el tratamiento con IMBRUVICA®. Modificaciones a la dosis de IMBRUVICA® no son requeridas.
Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continua de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de Farmacovigilancia: farmacovigilancia@digemid.minsa.gob.pe.
SOBREDOSIS:
No existe experiencia específica en el manejo de sobredosis con ibrutinib en los pacientes. Un sujeto sano experimentó incremento reversible de las enzimas hepáticas de grado 4 (AST y ALT) después de una dosis de 1680 mg. Monitorear cercanamente a los pacientes quienes ingieren más de la dosis recomendada y proporcionar tratamiento de soporte apropiado.
DESCRIPCIÓN: Ibrutinib es un inhibidor de la tirosina quinasa de Bruton (BTK). Es un sólido blanco a blanquecino con la fórmula empírica C25H24N6O2 y un peso molecular de 440.50 Ibrutinib es fácilmente soluble en dimetil sulfóxido, soluble en metanol y prácticamente insoluble en agua.
El nombre químico para ibrutinib es 1-[(3R)-3-[4-amino-3-(4-fenoxifenil)-1H-pirazolo[3,4d]pirimidin-1-i1]-1-piperidinil]-2-propen-1-una y tiene la siguiente estructura:
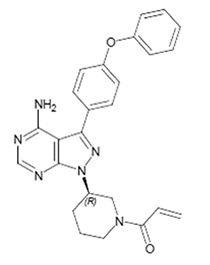
IMBRUVICA® (ibrutinib) cápsulas para administración oral es suministrado como cápsulas opacas blancas que contienen 140mg de ibrutinib como ingrediente activo. Cada cápsula blanca opaca está marcada con “ibr 140 mg” en tinta negra.
FARMACOLOGÍA CLÍNICA:
Mecanismo de acción: Ibrutinib es un inhibidor de moléculas pequeñas de la tirosina quinasa de Bruton (BTK). Ibrutinib forma un enlace covalente con un residuo de cisteína en el sitio activo de la BTK, conduciendo a la inhibición de la actividad enzimática de la BTK. La BTK es una molécula de señalización de las vías del receptor del antígeno de la célula B (BCR) y del receptor de la citoquina. El rol de la BTK en la señalización a través de los receptores de superficie de las células B produce la activación de vías necesarias para la adhesión, quimiotaxis y tráfico de las células B. Los estudios no clínicos muestran que ibrutinib inhibe la proliferación de las células B malignas y la supervivencia in vivo, así como la migración celular y la adhesión al sustrato in vitro.
Farmacodinamia: En pacientes con linfoma de células B recurrente se observó una ocupación > 90% del sitio activo de la BTK en células mononucleares de sangre periférica hasta 24 horas después de la dosis de ibrutinib ≥ 2.5 mg/kg/día (≥ 175 mg/día para un peso promedio de 70 kg).
En sujetos sanos, a una dosis única de 3 veces la dosis máxima recomendada (1680 mg), ibrutinib no prolongó el intervalo QT a ningún grado clínicamente relevante.
Farmacocinética:
Absorción: Ibrutinib se absorbe después de la administración oral, con una mediana del Tmáx de 1 a 2 horas. La exposición a ibrutinib se incrementa con dosis de hasta 840 mg. El AUC en el estado estacionario (media ± desviación estándar) observado en pacientes con 560 mg es 953 ± 705 ng.h/mL y en pacientes con 420 mg es 680 ± 517 ng.h/mL. La biodisponibilidad absoluta en condición de ayuno (n=8) fue 2.9% (IC al 90% = 2.1 – 3.9) y se duplicó cuando se combinó con los alimentos. La administración con los alimentos aumentó la Cmáx y el AUC de ibrutinib en aproximadamente 2 a 4 veces y 2 veces, respectivamente, en comparación con la administración de ibrutinib luego del ayuno durante la noche.
Distribución: La unión reversible de ibrutinib a la proteína plasmática humana in vitro fue 97.3% sin dependencia de la concentración en el rango de 50 a 1000 ng/mL. El volumen de distribución (Vd) fue 683 L y el volumen aparente de distribución en el estado estacionario (Vd,ss/F) fue aproximadamente 10000 L.
Metabolismo: El metabolismo es la principal vía de eliminación de ibrutinib. Es metabolizado a varios metabolitos principalmente por el citocromo P450, CYP3A y en menor grado por el CYP2D6. El metabolito activo, PCI-45227, es un metabolito dihidrodiol con una actividad inhibidora sobre la BTK aproximadamente 15 veces menor que ibrutinib. El rango de la proporción promedio entre el metabolito principal y el padre para PCI-45227 en el estado estacionario es 1 a 2.8.
Eliminación: La depuración intravenosa fue 62 y 76 L/h en condiciones de ayuno y alimentación, respectivamente. En línea con el alto efecto de primer paso, la depuración oral aparente es aproximadamente 2000 y 1000 L/h en condiciones de ayuno y alimentación, respectivamente. La vida media de ibrutinib es de 4 a 6 horas.
Ibrutinib, principalmente en forma de metabolitos, es eliminado principalmente por las heces. Luego de una administración única por vía oral de [14C]-ibrutinib radiomarcado en sujetos sanos, aproximadamente el 90% de la radioactividad fue excretada dentro de 168 horas, con la mayoría (80%) excretada en las heces y menos del 10% en la orina. El ibrutinib inalterado representó aproximadamente el 1% de la excreción del producto radiomarcado en las heces y no estuvo presente en la orina; el resto de la dosis fueron metabolitos.
Edad: En pacientes mayores (67 a 81 años de edad), existe una exposición prevista a ibrutinib del 14% mayor. No se justifica el ajuste de la dosis según la edad.
Género: El género no altera la depuración sistémica de ibrutinib.
Insuficiencia renal: Ibrutinib no es depurado significativamente por vía renal; la excreción urinaria de los metabolitos es < 10% de la dosis. La depuración de creatinina (CrCL) > 25 mL/min no tuvo influencia en la exposición a IMBRUVICA®. No existe información en pacientes con insuficiencia renal severa (CrCL < 25mL/min) o en pacientes con diálisis.
Insuficiencia hepática: Ibrutinib es metabolizado en el hígado. En un estudio de insuficiencia hepática, una dosis única de 140 mg de IMBRUVICA® se administró a sujetos sin cáncer. El AUC de ibrutinib se incrementó en 2.7, 2.8 y 9.8 veces, respectivamente, en sujetos con deterioro hepático leve (n=6), moderado (n=10) y severo (n = 8) comparado con sujetos con función hepática normal. La Cmáx de ibrutinib se incrementó en 5.2, 8.8 y 7.0 veces, respectivamente en sujetos con insuficiencia hepática leve, moderada y severa comparado con sujetos con función hepática normal (ver sección Uso en poblaciones específicas).
Interacciones con medicamentos:
Coadministración de ibrutinib con inhibidores del CYP3A: En un estudio de diseño secuencial de 18 voluntarios sanos en ayunas, una única dosis de 120 mg de IMBRUVICA® se administró solo el día 1 y una única dosis de 40 mg de IMBRUVICA® se administró el día 7 en combinación con 400 mg de ketoconazol (administrado una vez al día los días 4 – 9). Ketoconazol incrementó la Cmáx normalizada con la dosis y el AUC de ibrutinib en 29 veces y 24 veces, respectivamente. Simulaciones utilizando condiciones de ayuno indican que diltiazem y eritromicina inhibidores moderados del CYP3A, pueden incrementar el AUC de ibrutinib de 5 a 8 veces.
Coadministración de ibrutinib con inductores del CYP3A: Los datos farmacocinéticos de un estudio dedicado a la interacción de fármacos mostraron que la rifampicina (un inductor fuerte del CYP3A) disminuye la Cmáx y la AUC de ibrutinib por más de 13 y 10 veces. Las simulaciones usando modelos farmacocinéticos basados en fisiología (PBPK) sugirieron que un inductor moderado del CYP3A (efavirenz) puede disminuir hasta 3 veces el AUC de ibrutinib.
Coadministración de ibrutinib con sustratos del CYP: Estudios in vitro indicaron que es improbable que ibrutinib (I/Ki < 0.07 utilizando una Cmáx promedio a 560 mg) y PCI-45227 (I/Ki < 0.03) sean inhibidores de cualquiera de los CYPs principales a dosis clínicas. Tanto ibrutinib como el PCI-45227 son débiles inductores de las isoenzimas del CYP450 in vitro.
Coadministración de ibrutinib con substratos de transportadores: Los estudios in vitro indicaron que ibrutinib no es un sustrato de los transportadores de la Pgp (glucoproteína-P) o de la proteína de resistencia de cáncer de mama (BCRP) pero in vitro es un inhibidor de la P-gp y de la BCRP. Es improbable que ibrutinib sistémico sea un inhibidor de la P-gp a dosis clínicas ([I]1/Ki < 0.1) pero podría inhibir el BCRP. Ibrutinib puede tener un efecto sobre los sustratos de la P-gp o de la BCRP en el tracto gastrointestinal debido a las altas concentraciones locales después de una dosis oral. La co-administración de sustratos orales de la P-gp o de la BCRP con índice terapéutico estrecho (por ejemplo, digoxina, metotrexato) con IMBRUVICA® puede incrementar su concentración sanguínea.


