

LYRICA 75 MG, 150 MG
PREGABALINA
Cápsulas duras
2 Cápsulas duras, 75 Miligramos
14 Cápsulas duras, 75 Miligramos
28 Cápsulas duras, 75 Miligramos
2 Cápsulas duras, 150 Miligramos
14 Cápsulas duras, 150 Miligramos
28 Cápsulas duras, 150 Miligramos
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
LYRICA 75 mg Cápsula dura
Cada CÁPSULA DURA contiene: 75 mg de pregabalina.
Excipientes: Cada cápsula dura también contiene 8,25 mg de lactosa monohidrato.
LYRICA 150 mg Cápsula dura
Cada CÁPSULA DURA contiene: 150 mg de pregabalina.
Excipientes: Cada cápsula dura también contiene 16,50 mg de lactosa monohidrato
Para consultar la lista de excipientes ver Lista de excipientes.
FORMA FARMACÉUTICA: Cápsula dura.
LISTA DE EXCIPIENTES: Lactosa monohidrato, Almidón de maíz, Talco
INDICACIONES Y USOS: Dolor neuropático, dolor asociado con fibromialgia.
Precauciones relacionados con la indicación: El diagnóstico de fibriomialgia debería basarse cuidadosamente en los criterios internacionales tal como la clasificación de criterio (diagnóstico) del Colegio Americano de Reumatología y la droga debería ser administrada sólo cuando se haya realizado un diagnóstico definitivo.
FARMACOCINÉTICA
Concentración en sangre:
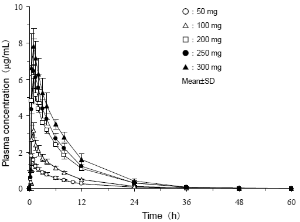
• Dosis simple: Una administración oral simple de 50, 100, 200, 250 y 300 mg de pregabalina en adultos saludables japoneses (seis sujetos por cada dosis) en ayunas, llegó a la Cmax en alrededor una hora después de la dosis y mostrando una T½ de alrededor seis horas. La Cmax y AUC0-8 se incrementó dependiendo de la dosis dentro de un rango de hasta 300 mg.
|
Dosis (mg) |
Cmax |
Tmax (h) |
AUC0-8 |
T1/2 (h) |
CL/F (L/h) |
Vd/F (L) |
Ae (%) |
|
50 |
2,03 (0,40) |
0,67 (0,26) |
10,7 (1,1) |
5,98 (0,65) |
4,72 (0,44) |
40,6 (4,9) |
83,9 (5,4) |
|
100 |
3,56 (0,67) |
0,75 (0,27) |
20,4 (1,3) |
5,66 (0,59) 4, |
93 (0,35) |
40,3 (6,4) |
95,0 (2,7) |
|
200 |
6,35 (0,73) |
1,00 (0,32) |
43,2 (3,0) |
5,93 (0,32) |
4,64 (0,32) |
39,7 (2,7) |
91,8 (2,6) |
|
250 |
7,18 (1,43) |
1,17 (0,52) |
49,2 (6,1) |
5,57 (0,72) |
5,15 (0,61) |
41,0 (3,8) |
95,6 (4,4) |
|
300 |
8,25 (1,36) |
1,08 (0,38) |
61,7 (6,3) |
5,80 (0,62) |
4,91 (0,52) |
40,9 (4,3) |
97,7 (7,3) |
|
Administración en ayunas; cada 6 sujetos; media (SD) Cmax: Máxima concentración de plasma Tmax: Tiempo para la máxima concentración de plasma AUC 0-8: Área bajo la curva del tiempo de concentración de plasma T1/2: Vida media de plasma CL/F: Aclaramiento total aparente Vd/F: Volumen de distribución aparente Ae (%): La tasa de excreción urinaria de la droga no cambiada hasta 60 horas después de la dosis simple |
|||||||
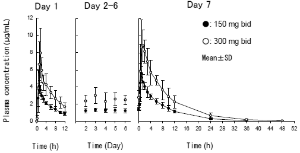
• Dosis múltiple: En una administración oral repetida de siete días de 150 o 300 mg de pregabalina dos veces al día a adultos saludables japoneses (7 sujetos por cada dosis), el estado estacionario llegó a las 24-48 horas después de la dosis y la T1/2 al día 7 fue de 5,95 y 6,31 horas respectivamente. La AUC0-12 al día 7 fue de 1,4 doble que al Día 1 de la dosis.
|
Cmax (µg/mL) |
Tmax (h) |
AUC0-12 (µg/mL) |
T1/2 (h) |
|||||
|
Día 1 |
Día 7 |
Día 1 |
Día 7 |
Día 1 |
Día 7 |
Día 1 |
Día 7 |
|
|
150 mg BID |
4,36 (0,68) |
6,24 (0,79) |
0,9 (0,4) |
0,9 (0,5) |
21,8 (1,7) |
30,7 (2,9) |
5,11 (0,75) |
5,95 (0,46) |
|
300 mg BID |
8,73 (2,52) |
10,5 (2,4) |
1,6 (1,1) |
1,6 (0,8) |
41,9 (7,4) |
58,7 (11,4) |
5,40 (0,94) |
6,31 (0,58) |
|
Media (SD), cada 8 sujetos |
||||||||
Efecto alimentario: Una administración oral simple de 150 mg de pregabalina en ayunas y después de comidas a 19 adultos saludables japoneses mostró una Cmax de 4,95 y 3,22 µgh/m, Tmax de 0,947 y 3,37 horas y AUC0-48 de 31,2 y 28,8 µgh/m/mL respectivamente. La administración después de las comidas bajó la Cmax por alrededor 35% y prolongó la Tmax por alrededor 2,4 horas, pero bajó la AUC0-48 sólo por alrededor de 8%.
Distribución: Una administración oral simple de 50, 100, 200, 250 y 300 mg de pregabalina en ayunas a japoneses adultos saludables (seis sujetos por cada dosis) mostró un volumen de distribución aparente (Vd/F) de alrededor 40 L.
La pregabalina se distribuyó en las células de la sangre y la tasa de concentración de sangre total a concentración de plasma fue de 0,76.
El enlace de proteína para pregabalina es insignificante a 0,1-20 µgh/mL (en estudio in vitro)
Metabolismo: La mayoría de pregabalina no se metaboliza. Alrededor de 99% de radioactividad recuperada en orina fue pregabalina sin cambios después de la administración de 100 mg de pregabalina-14C (107,9 µCi) en adultos saludables (seis extranjeros). El derivado N-metilado de pregabalina, el mayor metabolito de pregabalina encontrado en la orina, se contó en 0,9% de la dosis.
En el estudio in vitro, la administración de 159 µgh/mL de pregabalina (1 mM, alrededor de 10- veces más Cmax en el estado estacionario con una dosis de 600 mg/día) no mostró inhibición contra la CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 y CYP3A4.
Excreción: Una administración oral simple de 50, 100, 200, 250 y 300 mg de pregabalina en ayunas a japoneses adultos saludables (seis sujetos por cada dosis) mostró un CL/f de 4,64 a 5,15 L/h. La tasa de excreción urinaria en ese momento fue de 83,9 – 97,7%
Interacciones de la droga (información extranjera): Debido a que la pregabalina es predominantemente excretada sin cambios en la orina, experimenta un metabolismo insignificante en humanos y no enlaza a las proteínas plasmáticas, es poco probable que cause interacciones farmacocinéticas.
• Gabapentina: Las interacciones de pregabalina con gabapentina se examinaron en un estudio de dosis simple de 100 mg de pregabalina con 300 mg de gabapentina en 11 adultos saludables y un estudio de dosis repetida (en un intervalo de 8 horas) de 100 mg de pregabalina con 400 mg de gabapentina en 18 adultos saludables. Como resultado, las farmacocinéticas de la gabapentina no cambiaron por la co-administración con pregabalina en ambas, en el estudio de simple o dosis repetida. Además, la tasa de absorción de pregabalina fue ligeramente disminuida por la co-administración de gabapentina, pero su volumen de absorción no fue afectado.
• Interacciones de pregabalina con anticonceptivos orales (noretindrona, etinil estradiol): La co-administración de los siguientes de anticonceptivos orales una vez al día (combinación de 1mg de acetato de noretindrona y 0,035 mg de etinil estradiol) y 200 mg de pregabalina en cápsula tres veces al día a 16 mujeres adultas saludables, la administración concomitante de la Cmax de noretindrona con pregabalina fue comparable a aquella de sólo el anticonceptivo. La AUC0-24 incrementó al 16% con la administración concomitante con pregabalina. Así, la co-administración de pregabalina no afectó la farmacocinética de noretindrona. En relación al etinil estradiol, la Cmax y la AUC0-24 se incrementaron en un 5% y 14% respectivamente. Se concluyó que la co-administración de pregabalina no afectó la farmacocinética del etinil estradiol. Además, no hubo influencia en las concentraciones plasmáticas (presión mínima) de pregabalina al co-administrar un anticonceptivo oral.
• Lorazepam: Tras la co-adminsitración de 1 mg de lorazepam después de la administración oral múltiple de 300 mg de pregabalina dos veces al día a 12 sujetos saludables, la Cmax y la AUC0-8 de lorazepam se incrementó de 6% a 8% respectivamente comparado a sólo lorazepam. Así, la co-administración de pregabalina no afectó las farmacocinéticas de lorazepam. Por otra parte, la co-administración de lorazepam causó el 2% del aumento en la Cmáx y 1,8% de reducción de la AUC0-12 según se comparó con la topregabalina sola. La combinación de lorazepam no afectó las farmacocinéticas de pregabalina. La combinación de pregabalina y lorazepam redujo el rendimiento, tal como las veces de reacción y precisión, de la función motora gruesa y cognitiva en forma aditiva comparada con ambas lorazepam o pregabalina solas.
• Oxicodona: Tras la co-administración de 10 mg de oxicodona después de la administración oral múltiple de 300 mg de pregabalina dos veces al día a 12 sujetos saludables, la Cmax y AUC0-8 disminuyó un 1,1% y 9,5% respectivamente, según se comparó con oxicodona sola. Así, la co-administración de pregabalina no afectó las farmacocinéticas de oxicodona. Mientras, la Cmax de pregabalina con la administración concomitante de oxicodona se redujo en 4,5% según se comparó con pregabalina sola, la AUC0-12 fue comparable. La co-administración de oxicodona no afecto las farmacocinéticas de la pregabalina. La combinación de pregabalina y oxicodona redujo el rendimiento, tal como las veces de reacción y precisión, de la función motora gruesa y cognitiva en forma aditiva comparada con ambas oxicodona o pregabalina solas.
• Etanol: Tras la co-administración de 0,70 g/kg de etanol después de la administración oral múltiple de 300 mg de pregabalina dos veces al día a 13 sujetos saludables, la Cmax y AUC0-8 de etanol disminuyó en 8,9% y 9,6% respectivamente, según se comparó con etanol solo. Así, la co-administración de pregabalina no afectó las farmacocinéticas de etanol. Además, la Cmax y la AUC0-12 de pregabalina se incrementó en un 21% y 1% respectivamente, según se comparó con pregabalina sola, la cual no fue considerada como una diferencia clínicamente significativa. La combinación de pregabalina y etanol redujo el rendimiento, tal como las veces de reacción y precisión, de la función motora gruesa y cognitiva en forma aditiva comparada con ambas etanol o pregabalina solas.
• Fenitoína: Tras la administración oral múltiple de 200 mg de pregabalina 3 veces al día a 10 sujetos con epilepsia parcial bajo la dosis de mantenimiento de monoterapia de fenitoína, no hubo influencia en las concentraciones plasmáticas (presión mínima) de fenitoína. Además, la co-administración de fenitoína no afectó las farmacocinéticas de pregabalina
• Carbamazepina: Tras la administración oral múltiple de 200 mg de pregabalina 3 veces al día a 12 sujetos adultos con epilepsia parcial bajo la dosis de mantenimiento de la monoterapia con carbamazepina, no hubo influencia en las concentraciones de plasma (presión mínima) de carbamazepina y el metabolito, epóxido-10,11. Además, la co-administración de carbamazepina no afectó las farmacocinéticas de pregabalina.
• Ácido valproico: Tras la administración oral múltiple de 200 mg de pregabalina tres veces al día a 12 sujetos con epilepsia parcial bajo dosis de mantenimiento de monoterapia de valproato de sodio, no hubo influencia en las concentraciones de plasma (presión mínima) de valproato de sodio. Además, la co-administración de valproato de sodio no afectó las farmacocinéticas de pregabalina.
• Lamotrigine: Tras la administración oral múltiple de 200mg de pregabalina tres veces al día a 12 sujetos con epilepsia parcial bajo dosis de mantenimiento de monoterapia de lamotrigine, no hubo influencia en las concentraciones de plasma (presión mínima) de lamotrigine. Además, la co-administración de lamotrigine no afectó las farmacocinéticas de pregabalina.
Uso en ancianos: Una administración oral simple de 100 mg de pregabalina a 6 ancianos saludables japoneses de 67-78 años de edad mostró la Tmax de 1,4 horas y la T1/2 de 6,32 horas. Se confirmó que la AUC0-8 y la T1/2 tendieron a incrementarse y prolongarse ligeramente, comparadas a aquellas en sujetos no ancianos saludables quienes recibieron una administración oral simple de 100 mg de pregabalina.
|
Cmax (µg/mL) |
Tmax (h) |
AUC0-8 (µg h/mL) |
T1/2 (h) |
CL/F (L/h) |
|
|
Ancianos saludables |
3,24 (0,55) |
1,4 (0,5) |
26,6 (4,3) |
6,32 (0,82) |
3,82 (0,65) |
|
No ancianos saludables |
3,56 (0,67) |
0,75 (0,27) |
20,4 (1,3) |
5,66 (0,59) |
4,93 (0,35) |
|
Administración en ayunas; cada 6 sujetos; media (SD) |
|||||
Pacientes con daño renal:
• Información no japonesa: Una administración oral simple de 50 mg de pregabalina a 26 pacientes con diferentes funciones renales mostró una prolongada T1/2 y una incrementada AUC0-8 con una disminuida función renal. El CL/ F el aclaramiento renal (CLr) fueron proporcionales al aclaramiento de la creatinina.
|
Aclaramiento de creatinina |
Cmax (µg/mL) |
Tmax (h) |
AUC0-8 (µg h/mL) |
T1/2 (h) |
CL/F (mL/min) |
CLr (mL/min) |
|
>60 mL/min (n=11) |
1,86 (0,39) |
1,00 (0,224) |
15,9 (4,4) |
9,11 (2,83) |
56,5 (17,6) |
44,9 (23,6) |
|
>30 - <60 mL/min (n=7) |
1,53 (0,29) |
1,29 (0,393) |
28,2 (5,0) |
16,7 (4,1) |
30,6 (7,3) |
15,4 (7,7) |
|
>15 - <30 mL/min (n=7) |
1,90 (0,62) |
1,93 (1,48) |
52,3 (11,7) |
25,0 (6,7) |
16,7 (3,9) |
9,23 (3,37) |
|
<15 mL/min (n=1) |
1,69 |
1,00 |
101 |
48,7 |
8,30 |
4,30 |
|
Dosis: 50mg (dosis simple; Media (SD) CLr: Aclaramiento renal |
||||||
• Información japonesa (farmacocinética poblacional): Como resultado de los análisis farmacocinético poblacional, se estableció un modelo de un compartimiento con absorción de primer orden para describir la información farmacocinética de 838 sujetos (incluyendo 474 sujetos japoneses: 70 voluntarios sanos, 26 pacientes con neuralgia postherpética, 154 DPN con dolor asociado a neuropatía periférica diabética y 224 pacientes con fibromialgia). El aclaramiento de creatinina e IBW fueron seleccionados como co-variables de CL/F y BMI, se seleccionó el peso corporal ideal, género y edad como co-variables de Vd/F. CLcr para CL/F se consideró un factor significativo que afectaba las farmacocinéticas de la pregabalina. En pacientes con daño renal, la CL/F disminuye en correlación con el CLcr, por lo tanto, se debe ajustar la dosis de acuerdo al CLcr.
El estimado estado estacionario de AUC (AUC0-8, SS) en pacientes japoneses con dolor asociado a la neuropatía periférica diabética (DPN) con CLcr <60 mL/min tras la pregabalina 150 mg dos veces al día (300 mg/día) fue similar a aquel en pacientes con DPN con CLcr >60 mL/min tras pregabalina 300 mg dos veces al día (600 mg/día). El aclaramiento de pregabalina (CL/F) en pacientes con CLcr <60 mL/min fue la mitad de CL/F en pacientes con CLcr >60 mL/min.
|
Aclaramiento de creatinina |
Dosis (mg/día) |
AUC0-8 (µg h/mL) |
CL/F (mL/min) |
|
≥60 mL/min (n=31) |
600 |
86,1 (27,8) |
63,6 (18,5) |
|
≥30 - <60 mL/min (n=14) |
300 |
85,7 (22,6) |
31,1 (8,11) |
|
Media (SD) |
|||
Pacientes de hemodiálisis (información no japonesa): Una administración oral simple de 50mg de pregabalina a 12 pacientes que se someten a hemodiálisis mostró que la hemodiálisis cada 4 horas bajó la concentración de pregabalina en plasma a alrededor de 50%. El aclaramiento de hemodiálisis fue de 192 mL/min.
CONTRAINDICACIONES
Esta droga está contraindicada en los siguientes pacientes:
• Pacientes con historia de hipersensibilidad a algunos de los ingredientes del producto.
PRECAUCIONES RELACIONADAS CON EL USO
Precauciones relacionadas con la dispensación: En caso de empaques prensados (PTP), instruir al paciente para que retire la droga del empaque antes de su uso. [Se ha reportado que si el PTP está inflado, los bordes filosos del empaque pueden perforar el esófago, resultado en complicaciones serias tal como mediastinitis.]
Otras precauciones:
• En análisis de 199 estudios clínicos placebo-controlados de varias drogas antiepilépticas en pacientes con epilepsia o trastornos psiquiátricos, etc., se estimó que la diferencia de riesgo de pacientes en el grupo de tratamiento de la droga quienes experimentaron ideas o comportamiento suicida comparado a pacientes del grupo de placebo es 1,9 por 1000 (95% CI: 0,6 – 3,9). Este análisis mostró que un riesgo de idea o comportamiento suicida en pacientes randomizados en una de las drogas antiepilépticas (0,43%) fue aproximadamente dos veces comparados a pacientes randomizados con placebo (0,24%). De acuerdo con el análisis de subgrupo, también hubo un estimado de 2,4 por 1000 eventos suicidas en pacientes con epilepsia en el grupo de tratamiento con la droga que con el grupo con placebo*.
* Las indicaciones de esta droga aprobada son dolor neuropático y Dolor asociado con fibromialgia, aunque ha sido aprobado como fármaco antiepilético en el extranjero.
• Un estudio de carcinogenicidad de dos años en ratones tratados con la dosis de pregabalina la cual era equivalente al nivel de exposición más de 6 veces más altas que el nivel de exposición de la media de seres humanos de la máxima dosis clínica en humanos mostró una incidencia aumentada de dosis-dependiente de hemangiosarcoma.
• Se vio un incremento en la incidencia de la atrofia retinal comúnmente observada en ratas albinas en estudios de carcinogenicidad de dos años en ratas tratadas con pregabalina en exposiciones a ≥5 veces la media de la exposición en seres humanos en la dosis clínica máxima recomendada (párrafo siguiente). El estudio de distribución tisular en ratas reveló que la radiación derivada de pregabalina 14C se eliminó más lentamente en el lente que en la sangre y casi todos los otros tejidos, sin embargo, en los estudios de toxicidad de dosis repetidas en ratas no se observó ningún efecto en los lentes en ambos 13- y 52 semanas. En un estudio placebo-controlado de 13 a 16 semanas con pacientes con neuralgia postherpética (3 estudios), la incidencia de las reacciones adversas fue 10,6% en el grupo de pregabalina (300 – 600 mg/día), mientras que en el grupo placebo fue 3.8%. En un estudio de administración a largo plazo con pacientes con neuralgia postherpética, la incidencia de las reacciones adversas observadas en los pacientes tratados con pregabalina fue 10%. En el estudio placebo-controlado de 16 semanas con fibromialgia conducido, la incidencia de reacciones adversas fue de 9.2% en el grupo de pregabalina (300 – 450mg/día) mientras en el grupo del lugar fue 2,8%. En el estudio de administración a largo plazo con fibromialgia conducido en Japón, la incidencia de reacciones adversas observados en pacientes tratados con pregabalina fue de 9,4%.
• En el estudio de fertilidad y desarrollo embriónico temprano en ratas machos, la pregabalina indujo la toxicidad reproductiva masculina a exposiciones >28 veces la exposición media en humanos en la dosis clínica máxima recomendada.
La revisión de 199 estudios clínicos de 11 drogas antiepilépticas han demostrado que los pacientes que reciban medicamentos antiepilépticos tienen casi dos veces el riesgo del comportamiento o pensamiento suicida (0,43 por ciento) comparado a los pacientes que recibían placebo (0,24 por ciento), Esta diferencia fue cerca de un caso adicional de pensamiento o comportamiento suicida por cada 500 pacientes tratados con drogas antiepilépticas versus placebo.
Cuatro de los pacientes que fueron seleccionados al azar para recibir una de las drogas antiepilépticas cometieron suicido, mientras que en el grupo placebo no hubo ningún paciente que lo hiciera.
Se recuerda a los médicos que deben aconsejar e instruir a sus pacientes, familiares y personal auxiliar que deben estar atentos para identificar tempranamente los signos y/o síntomas de pensamientos y/o comportamientos suicida asi como los cambios de humor.
Los pacientes y/o familiares deben ponerse en contacto con su médico, si perciben cambios de humor en el paciente o que presenten comportamiento y/o pensamiento suicida.
REACCIONES ADVERSAS
La neuralgia posherpética: En un estudio japonés de respuesta a la dosis, a largo plazo, estudio Fase II, estudio Fase III y estudio a largo plazo, se observaron reacciones adversas en 1084 pacientes (64,5%) de 1680 pacientes quienes habían recibido pregabalina 75-600 mg/dia BID o TID. Las reacciones adversas más comunes observadas fueron mareo, 393 pacientes (23,4%); somnolencia, 267 pacientes (15,9%); y edema, 179 pacientes (10,7%). (Tabulación de los resultados del estudio hasta el momento de la aprobación).
Dolor asociado con la neuropatía periférica diabética: En un estudio comparativo doble-ciego japonés y estudio a largo plazo, en pacientes con dolor asociado a neuropatía periférica diabética se observaron reacciones adversas en 199 pacientes (65,9%) de 302 pacientes quienes habían recibido pregabalina de 150 – 600 mg/día BID. Las reacciones adversas más comunes observadas fueron somnolencia 74 pacientes (24,5%), mareo 68 pacientes (22,5%) y edemas 52 pacientes (17,2%). (Tabulación de los resultados del estudio al momento de la aprobación).
Dolor asociado con lesiones de la medula espinal, dolor tras un accidente cerebro vascular y el dolor de esclerosis múltiple: En un estudio comparativo global doble-ciego en pacientes con dolor asociado con lesiones a la medula espinal y en un estudio japonés a largo plazo en pacientes con dolor asociado con lesiones en la medula espinal, dolor tras un accidente cerebro vascular y dolor de esclerosis múltiple, las reacciones adversas fueron observadas en 165 pacientes (76,7%) de 215 pacientes quienes habían recibido pregabalina 150 – 600 mg/dia dos veces al día. Las reacciones adversas más frecuentes observadas fueron somnolencia en 87 pacientes (40,5%), mareo en 43 pacientes (20,0%), y edema en 40 pacientes (18,6%). La tabulación de los resultados hasta el momento de la aprobación)
Fibromialgia: En un estudio comparativo doble-ciego japonés y a largo plazo, en pacientes con fibromialgia se observaron reacciones adversas en 295 pacientes (82,9%) de 356 pacientes quienes habían recibido pregabalina 300 - 450 mg/día BID. Las reacciones adversas más comunes observadas fueron somnolencia en 141 pacientes (39,6%), mareo 98 pacientes (27,5%) e incremento de peso 56 pacientes (15,7%). (Tabulación de los resultados del estudio al momento de la aprobación)
Reacciones adversas clínicamente significativas
• Mareo (sobre el 20%), somnolencia (sobre el 20%) y pérdida del conocimiento (menor de 0,3%): Debido a que pueden ocurrir fracturas relacionadas con las caídas asociadas a la pérdida de conciencia, somnolencia y mareo, se deben observa a los pacientes muy de cerca. Si se observaran anormalidades, se deberá descontinuar la administración o reducir la dosis y tomar medidas terapéuticas apropiadas.
• Insuficiencia cardiaca (menor a 0,3%), edema pulmonar (incidencia desconocida*): Debido a que se ha reportado insuficiencia cardiaca y edema pulmonar, particularmente en pacientes con enfermedad cardiovascular, se deberá observar muy de cerca a los pacientes en riesgo de desarrollar insuficiencia cardiaca. Si se observaran anormalidades, se deberá descontinuar la administración y tomar las medidas terapéuticas necesarias.
• La rabdomiólisis (incidencia desconocida*): Dado que puede aparecer rabdomiólisis se deberá observar al paciente muy de cerca y cuando se reconozca mialgia, debilidad, aumento de CK (CPK), aumento de mioglobina en sangre/orina etc. Se deberá descontinuar la administración y tomar las medidas apropiadas. Adicionalmente, se deberá tener precaución en caso ocurriera un daño renal agudo causado por la rabdomiólisis.
• Falla renal (menor a 0,1%): Debido a que se ha reportado falla renal, deberá descontinuarse la administración si ésta se desarrolla, asimismo, se deberá tomar las medidas terapéuticas apropiadas.
• Angioedema (incidencia desconocida*): Pueden ocurrir reacciones de hipersensibilidad tal como la angioedema en un paciente que recibe la droga, si se observara cualquier anormalidad, se deberá descontinuar la administración inmediatamente y tomar las medidas terapéuticas apropiadas.
• Hipoglicemia (menor del 0,3%): Debido a que puede aparecer la hipoglicemia en un paciente que recibe la droga, se deberá descontinuar la administración y tomar las medidas apropiadas cuando se reconozca algún hallazgo anormal de hipoglicemia, tales como sensación de debilidad, malestar, sudor frío, temblor y alteraciones en la conciencia.
• Neumonía intersticial (incidencia desconocida*): Debido a que la neumonía intersticial puede aparecer en un paciente que recibe la droga, las condiciones clínicas tales como tos, disnea, fiebre se deben monitorear cuidadosamente. Si se reconociera cualquier hallazgo anormal, se deberán realizar el diagnóstico y medidas terapéuticas como Rayos X de tórax y Scan CT de tórax. Se deberá descontinuar la administración, realizar el diagnóstico apropiado y tomar las medidas terapéuticas, tal como la administración de corticosteroides suprarrenales.
• Golpes (incidencia desconocida*) y síntomas anafilactoides (menos de 0,1%): Debido a que pueden aparecer golpes y síntomas anafilactoides en pacientes que reciben la droga, éstos deben ser monitoreados cuidadosamente y si se observaran anormalidades, se deberá descontinuar la administración inmediatamente y tomar las medidas terapéuticas apropiadas.
• Síndrome oculomucocutaáneo (Síndrome Stevens-Johnson) (incidencia desconocida*) eritema multiforme (incidencia desconocida*): Debido a que puede aparecer síndrome oculomucocutáneo y eritema multiforme en un paciente que recibe la droga, éstos deben ser monitoreados cuidadosamente y si se observaran anormalidades, se deberá descontinuar la administración inmediatamente y tomar las medidas terapéuticas apropiadas.
* Clasificado como incidencia desconocida dado que son casos espontáneos y/o reportados en otros países
Otras reacciones adversas
Si se observan las siguientes reacciones adversas a la droga, se deberán implementar las medidas apropiadas tal como reducción de la dosis o descontinuación.
|
≥1% |
≥0,3% <1% |
Menor de 0,3% |
Incidencia desconocida * |
|
|
Trastornos de sangre y sistema linfático |
Neutropenia, leucopenia |
Trombocitopenia |
||
|
Trastornos de metabolismo y nutrición |
Anorexia, aumento del apetito, hiperlipidemia |
Hiperglicemia |
||
|
Trastornos psiquiátricos |
Insomnio, confusión, desorientación, euforia, alucinación, sueños anormales |
Depresión, inquietud, cambios de humor, estado de ánimo deprimido, apatía, ansiedad, pérdida de la libido, trastorno del sueño, pensamientos anormales |
Despersonalización, anorgasmia, agitación, dificultad para encontrar las palabras, incremento de la libido, ataque de pánico, desinhibición |
|
|
Trastornos del sistema nervioso |
Mareo, dolor de cabeza, trastorno del equilibrio, ataxia |
Temblor, alteración de la atención, letargia, deterioro de la memoria, amnesia, disartria, parestesia, hipoanestesia, coordinación anormal |
Sedación, trastorno cognitivo, mioclono, hiporeflexia, disquinesia, hiperactividad psicomotora, mareo postural, hiperestesia, ageusia, sensación de ardor, síncope, deficiencia mental, trastorno del habla |
Estupor, parosmia, disgrafía |
|
Trastornos oculares |
Visión borrosa, diplopía, reducción de la agudeza visual |
Alteración de la visión, hemorragia retiniana |
Defecto del campo visual, hinchazón de los ojos, dolor de los ojos, astenopia, incremento del lagrimeo, fotopsia, estrabismo, ojo seco |
Irritación ocular, midriasis, oscilopsia, percepción de la profundidad visual alterada, brillo visual, nistagmo, queratitis |
|
Trastornos del oído y laberintitis |
Vértigo |
Tinitus |
Hiperacusia |
|
|
Trastornos cardiacos |
Palpitaciones |
Bloqueo auriculoventricular de primer grado, taquicardia, arritmia sinusal, bradicardia sinusal, extrasístoles ventriculares |
Taquicardia sinusal |
|
|
Trastornos vasculares |
Hipertensión, sofocos |
Hipotensión |
||
|
Trastornos respiratorios, toráxicos y mediastinales |
Disnea |
Nasofaringitis, tos, ronquidos, epistaxis, rinitis |
Sequedad nasal, congestión nasal, opresión en la garganta |
|
|
Trastornos gastrointestinales |
Estreñimiento, nausea, diarrea, vómito, dolor abdominal |
Distensión abdominal, flatulencia, dispepsia, malestar estomacal, estomatitis |
Hipersecreción salival, enfermedad por reflujo gastroesofágico, pancreatitis, inflamación de la lengua |
Ascitis, disfagia |
|
Trastornos de la piel y tejido subcutáneo |
Erupción |
Prurito, edema periorbital, eccema |
Sudoración, sudor frío, urticaria, alopecia |
Erupción papular, alopecia |
|
Trastornos musculoesqueléticos y tejido contector |
Hinchazón de las articulaciones, debilidad muscular, espasmos musculares, dolor extremo, dolor de espalda |
Mialgia, sensación de pesadez, artralgia, rigidez muscular |
||
|
Trastornos renal y urinario |
Incontinencia urinaria |
Disuria |
Oliguria, retención urinaria |
|
|
Trastornos del sistema reproductivo y pecho |
Dolor de pecho, disfunción eréctil, ginecomastia |
Retraso de la eyaculación, disfunción sexual, amenorrea, secreción de las mamas, dismenorrea, hipertrofia de las mamas |
||
|
Trastornos generales y condiciones del lugar de la administración |
Edema, sed, fatiga, trastorno de la marcha, edema facial, sensación anormal |
Astenia, edema con fóvea, dolor, malestar general, dolor en el pecho |
Fiebre, sensación de frío, escalofríos, irritabilidad, sensación de embriaguez |
Opresión en el pecho |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Caída |
|||
|
Trastornos hepatobiliares |
Función hepática anormal |
|||
|
Investigaciones |
Incremento de peso |
Aumento de la creatina fosfoquinasa en sangre, aumento de la creatinina en sangre, aumento de la amilasa en sangre, incremento de la AST (TGO), incremento de la ALT (TGP) |
Disminución de peso, aumento del ácido úrico en sangre |
Reducción del potasio en sangre |
|
* La incidencia es desconocida porque la información de estas reacciones adversas incluyeron información clínica extranjera e información de post-marketing de las indicaciones no aprobadas en Japón. |
||||
Uso en ancianos: Dado que los pacientes de edad avanzada son más propensos a padecer disfunción renal, sus dosis e intervalos de dosis deberían ajustarse basándose en el aclaramiento de la creatinina y administrarse con cuidado (ver Precauciones relacionadas a la dosis y administración, Administración cuidadosa y Farmacocinética).
Adicionalmente, se han reportado fracturas relacionadas con las caídas en pacientes ancianos asociados con la pérdida del conocimiento, somnolencia y mareo. Por lo tanto, se requiere especial precaución cuando se administre esta droga (ver Precauciones importantes y Reacciones adversas clínicamente significativas).
Uso durante el embarazo, parto o lactancia:
• Embarazo: Esta droga no debe usarse en mujeres embarazadas o mujeres que posiblemente puedan embarazarse a menos que los beneficios terapéuticos esperados tengan mayor peso que los posibles riesgos asociados con el tratamiento. [No se ha establecido la seguridad de esta droga en mujeres embarazadas. En experimento en animales, se reportaron anormalidades fetales (bajo peso, aumento en la incidencia del edema localizado, mutación esquelética, retraso de la osificación, etc.) influencia en niños recién nacidos (bajo peso, tasa de supervivencia reducida, reacción de sobresalto auditivo reducido, retraso en el desarrollo, influencia en la capacidad reproductiva, etc.).]
• Madres que dan de lactar: Se deberá descontinuar la lactancia durante la administración de esta droga [Aunque no se sabe si la droga se excreta en la leche materna de los humanos, porque sí está presente en la leche de las ratas.]
Uso en niños, etc.: No se ha establecido la seguridad de esta droga en niños de bajo peso al nacer, neonatos, infantes, niños infantes o niños. (No hay estudios clínicos japoneses con experiencia en uso clínico) [Las ratas jóvenes son más sensibles a esta droga. En la exposición de las ratas jóvenes equivalentes a la exposición media en humanos a la dosis clínica máxima recomendada (600 mg/día), hubo evidencia de signos clínicos de CNS de hiperactividad y bruxismo y algunos cambios en el crecimiento (incremento en el peso corporal de supresión transitoria). Se observó una reducción de la respuesta de sobresalto acústico en ratas después de una exposición de >2 veces la exposición terapéutica en seres humanos y el intervalo de anestro prolongado después de la exposición de 5 veces más a la exposición terapéutica humana.]
INCOMPATIBILIDADES: No aplicable.
ADVERTENCIAS: El uso de este medicamento puede producir o incrementar pensamientos y/o comportamientos suicidas.
Los profesionales de la salud prescriptores o quienes están al cuidado de los pacientes asi como sus familiares deben estar alertas con la finalidad de identificar tempranamente la aparición o el empeoramiento o comportamiento suicida o cualquier cambio inusual en el humor o comportamiento.
Estos efectos adversos pueden producirse en los pacientes que reciben tratamiento con estos medicamentos para el tratamiento de la epilepsia, cefalea, migraña, dolor neuropático o cualquier otra indicación durante el tratamiento.
PRECAUCIONES
Administración cuidadosa (esta droga deberá administrarse con cuidado en los siguientes pacientes
• Pacientes con daño renal (ver Precauciones relacionadas a la dosis y administración y Farmacocinética).
• Pacientes con insuficiencia cardíaca congestiva grave [Insuficiencia cardiaca congestiva puede aparecer en enfermedades cardiovasculares (ver Reacciones adversas”)].
• Ancianos (ver Precauciones importantes y Uso en ancianos).
• Pacientes con historia de angioedema (ver Reacciones adversas).
Precauciones importantes:
• Se observaron mareo, somnolencia y pérdida del conocimiento durante el tratamiento de la droga, llevando a algunos accidentes de tránsito. Se debe aconsejar a los pacientes que no manejen maquinaria peligrosa, tal como conducir un automóvil durante el tratamiento. Se han reportado fracturas relacionadas con caídas causadas por estos síntomas, especialmente en pacientes ancianos. Por lo tanto, se debería advertirles que sean cautelosos.
• Después de una descontinuación repentina de la droga, se puede observar, síntomas tales como insomnio, náusea, dolor de cabeza, diarrea, ansiedad e hiperhidrosis. Si el tratamiento de la droga tiene que ser descontinuada, la dosis debe reducirse gradualmente en un mínimo de una semana.
• Se puede ganar peso con el tratamiento con esta droga, y por lo tanto los pacientes deberían ser monitoreados en caso de producirse obesidad o apareciera algún signo de obesidad. Se deberán tomar medidas apropiadas tal como una dieta adecuada y realizar ejercicios. Se deberá controlar el peso periódicamente porque éste se puede incrementar al aumentar la dosis o con la administración de largo plazo en particular.
• Dado que esta droga puede causar problemas visuales tales como la ambliopía, visión anormal, visión borrosa y diplopía, los doctores deben conversar con sus pacientes en caso de cualquier problema visual. Si apareciera cualquier signo de anormalidades, se deberán tomar medidas apropiadas (ver Otras precauciones).
El tratamiento para el dolor neuropático periférico con pregabalina no constituye una terapia causal sino una terapia sintomática; en consecuencia, el médico deberá también realizar el diagnóstico y tratamiento de la enfermedad la cual es considerada la causa del dolor.
Interacciones de la droga
Precauciones para la co-administración (esta droga debe ser administrada con cuidado cuando se co-administre con las siguientes drogas)
|
Drogas |
Signos, síntomas y tratamiento |
Mecanismo y factores de riesgo |
|
Medicamentos depresores del SNC Analgésicos opiáceos |
Ha habido reportes de falla respiratoria y coma |
Mecanismo no claro |
|
Oxycodona Lorazepam Alcohol (bebida) |
La pregabalina parece ser como un aditivo en el deterioro de la función cognitiva y motora causada por estas drogas |
Debido al efecto aditivo |
|
Drogas que desarrollen angioedemas (inhibidores de la emzima angiotensina, etc.) |
Los pacientes que toman estas drogas, las cuales son conocidas por tener una relación con las angioedemas, puede incrementar los riesgos de desarrollo de angioedemas (edema facial, edema bucal, edema cervical, etc.) |
Mecanismo no claro |
|
Drogas que desarrollen edema periférico (fármacos tiazolidina, etc) |
Los riesgos para desarrollar edema periférico pueden incrementarse debido a la co-administración de la pregabalina y a fármacos tiazolidina. Como los fármacos tiazolidina pueden causar incremento de peso y/o retención de líquidos, posiblemente lleve a exacerbar la falla cardiaca, tenga precaución cuando se co-administre pregabalina y estos agentes |
Mecanismo no claro |
DOSIS Y ADMINISTRACIÓN
Para el dolor neuropático: La dosis usual para adultos empieza con 150 mg/día de pregabalina dos veces al día y deberá aumentarse gradualmente a 300 mg/día en una semana o más y deberá administrarse oralmente dos veces al día. Se deberá ajustar la dosis, dependiendo de la edad o síntomas. Sin embargo, la dosis máxima diaria no deberá ser más allá de 600 mg, y deberá administrarse oralmente dos veces al día.
Para el dolor asociado con la fibromialgia: La dosis usual para adultos empieza con 150 mg/día de pregabalina dos veces al día y deberá aumentarse gradualmente a 300 mg/día en una semana o más y deberá mantenerse en 300-450 mg /día según sea necesario. La dosis deberá ajustarse, dependiendo de la edad o síntomas. Sin embargo, la dosis máxima diaria no deberá ser más allá de 450 mg y deberá administrarse oralmente dos veces al día.
Precauciones relacionados con la dosis y administración:
• Si se tiene que descontinuar el tratamiento con esta droga, la dosis debe disminuirse gradualmente sobre un mínimo de una semana (ver Precauciones importantes).
• La pregabalina se excreta en la orina, principalmente en una forma no-metabolizada. En un paciente con daño renal, la concentración de plasma de pregabalina puede ser mayor, lo que aumenta la probabilidad de reacciones adversas a los medicamentos. Por lo tanto, las condiciones del paciente deberán ser monitoreadas cuidadosamente y se deberá tener mucha cautela cuando se use pregabalina con tal paciente. La dosis y los intervalos de dosis en pacientes con daño renal deben ser ajustados de acuerdo al aclaramiento de la creatinina, según se indica en la tabla de abajo. Para los pacientes que se someten a hemodiálisis, además de la dosis diaria de pregabalina basada en el aclaramiento de creatinina de cada paciente, se deberá dar una dosis adicional inmediatamente después de cada tratamiento de hemodiálisis. Cuando se han puesto varias dosis, empezar desde la dosis más baja, y si se confirma la tolerabilidad y la respuesta es inadecuada, incrementar la dosis. La dosis y administración que se muestra aquí se basa en resultados de simulación, por lo tanto se debe ajustar la dosis y administración por cada paciente con un cuidadoso seguimiento (ver Farmacocinética).
Para el dolor neuropático
|
Aclaramiento de creatinina (mL/min) |
≥60 |
≥30 - <60 |
≥15 - <30 |
<15 |
Dosis suplementaria Posthemodiálisis* |
|
Dosis diaria |
150-600 mg |
75 – 300 mg |
25 – 150 mg |
25 – 75 mg |
- |
|
Dosis inicial |
75 mg BID |
25 mg TID o 75 mg QD |
25 mg QD o BID o 50 MG QD |
25 QD |
25 o 50 mg |
|
Dosis de mantenimiento |
150 mg BID |
50 mg TID o 75 mg BID |
75 mg QD |
25 o 50 mg QD |
50 o 75 mg |
|
Dosis máxima |
300 mg BID |
100 mg TID o 150 mg BID |
75 mg BID o 150 mg QD |
75 mg QD |
100 o 150 mg |
|
* Basado en los resultados de una simulación cuando un paciente tiene una hemodiálisis de 4 horas cada 2 días, 6 horas después de la administración. |
|||||
Para el dolor asociado a fibromialgia
|
Aclaramiento de creatinina (mL/min) |
≥60 |
≥30 - <60 |
≥15 - <30 |
<15 |
Dosis suplementaria Posthemodiálisis* |
|
Dosis diaria |
150-450 mg |
75-225 mg |
25 – 150 mg |
25 – 75 mg |
- |
|
Dosis inicial |
75 mg BID |
25 mg TID o 75 mg QD |
25 mg QD o BID o 50 MG QD |
25 QD |
25 o 50 mg |
|
Dosis de mantenimiento |
150 mg BID |
50 mg TID o 75 mg BID |
75 mg QD |
25 o 50 mg QD |
50 o 75 mg |
|
Dosis de mantenimiento (Dosis máxima) |
225 mg BID |
75 mg TID |
100 mg o 125 mg QD o 75 mg BID |
50 mg o 75 mg QD |
75 o 100 mg |
|
* Basado en los resultados de una simulación cuando un paciente tiene una hemodiálisis de 4 horas cada 2 días, 6 horas después de la administración. |
|||||
VÍA DE ADMINISTRACIÓN: Vía oral.
SOBREDOSIS
Síntomas: Se reportaron casos de sobredosis hasta 15 g, los síntomas observados más comunes reportados por sobredosis fue trastorno afectivo, somnolencia, estado de confusión, depresión, agitación e inquietud.
Intervención: Se deberá realizar el tratamiento sintomático. Debido a que esta droga se elimina por hemodiálisis, se debería considerar realizar una hemodiálisis, dependiendo de la severidad de los síntomas de inicio (ver Farmacocinética).
FARMACOLOGÍA
Efecto analgésico: En estudios en animales, la pregabalina no previene comportamientos en respuesta al dolor no adictivo agudo, pero inhibe el dolor neuropático asociado con las lesiones de nervios periféricos, diabetes y dolor musculoesquelético crónico. Además la pregabalina también tiene efectos analgésicos y dolores espontáneos/modelos de hiperalgesia provocados por productos químicos, inflamación o lesiones del tejido:
• Efecto antialodínico* en el modelo (CCI) de la lesión crónica constrictiva*: En el modelo CCI, la pregabalina inhibió ambas, la alodinia estática y dinámica en ratas.
• Efecto antilodínico en el modelo (SNL) de la ligadura del nervio espinal: En el modelo SNL, la pregabalina inhibió ambas, la alodinia estática y dinámica en ratas.
• Efecto antilodínico en el modelo de diabetes STZ: En el modelo de diabetes STZ, la pregabalina inhibió ambas, la alodinia estática y dinámica en ratas.
• Efecto antilodínico en el modelo del dolor tras una lesión en la medula espinal: La pregabalina inhibió la alodinia estática ocurrida en el ratón en el modelo de la lesión de la medula espinal bajo peso.
• Efecto antilodínico en el modelo de dolor musculoesquelético crónico: En el modelo de dolor musculoesquelético crónico, la pregabalina inhibió la alodinia estática y dinámica en ratas.
• Efecto analgésico en dolor espontáneo en la prueba de lesiones plantares de formalina: Para las respuestas relacionadas con dolor bifásicos ocasionados por la administración de formalina en los plantares de las ratas, la pregabalina inhibió la fase retrasada en la cual se consideró involucrada a la sensibilización central.
* La alodinia táctil es un dolor provocado normalmente por la estimulación táctil inocua y es clasificada como alodinia estática (provocada por una estimulación ligera puntuar sobre la piel) o dinámica (por un golpe ligero sobre la piel).
Mecanismo de acción: Se mostró que la pregabalina inhibe la liberación de neurotransmisores como el ácido glutamínico a través de la reducción de la expresión del canal de calcio en la superficie de la célula y/o afluencia de calcio tras su enlace a la subunidad α2δ, la cual juega un rol auxiliar para la función del canal de calcio de voltaje dependiente en el sistema nervioso central. Además, se sugiere que las actividades anti-adictivas de la pregabalina pueden también ser mediadas a través de las interacciones con las vías descendientes noradrenérgicas y serotonérgicas que modulan la transmisión del dolor.
FORMAS DE PRESENTACIÓN Y CONCENTRACIONES
LYRICA Cápsulas 75 mg: 2, 14 y 28 cápsulas duras.
LYRICA Cápsulas 150 mg: 2, 14 y 28 cápsulas duras.
CONDICIONES DE ALMACENAMIENTO: Ver condiciones de almacenamiento en la caja
Fabricado por:
Pfizer Manufacturing Deutchsland GmbH – Alemania
Importado por:
PFIZER S.A.
Av. Javier Prado Este N° 6230 – 2 do Piso
Lima 12- Perú
Teléfono: 615-2100, Fax: 615-2106
LLD basado en Ficha técnica Japón 28.02.2013 V.3


