

XOLAIR
OMALIZUMAB
Vial
1 Vial de polvo , 1 Ampollas inyectables , 150 Miligramos
DESCRIPCIÓN Y COMPOSICIÓN:
Formas farmacéuticas: Vial de polvo y ampolla de disolvente para solución inyectable.
Un vial de XOLAIR® 150 mg contiene: 150 mg de omalizumab. La solución reconstituida de XOLAIR® contiene 125 mg/ml de omalizumab (150 mg en 1,2 ml).
Sustancia farmacéutica: El omalizumab es un anticuerpo monoclonal humanizado obtenido de una línea celular mamífera.
Es posible que algunas dosis y formas farmacéuticas no estén disponibles en todos los países.
Excipientes:
• Vial de polvo y ampolla de disolvente para solución inyectable:
— Vial de polvo: Sacarosa, L-histidina, clorhidrato de L-histidina monohidratado, polisorbato 20.
— Ampolla de disolvente: Agua para preparaciones inyectables.
INDICACIONES:
Asma alérgica: XOLAIR® (omalizumab) está indicado para el tratamiento de los adultos, adolescentes y niños (de 6 a < 12 años de edad).
El tratamiento con XOLAIR® deberá ser considerado únicamente para pacientes con asma mediada de forma convincente por IgE (inmunoglobulina E).
• Adultos y adolescentes (a partir de 12 años de edad): XOLAIR® está indicado para mejorar el control del asma cuando se administra como tratamiento adicional en pacientes con asma alérgica grave persistente que presentan test cutáneo positivo o reactividad in vitro a aeroalergenos perennes y con función pulmonar reducida (FEV1 <80%) así como, síntomas frecuentes durante el día o despertares por la noche y que han presentado múltiples exacerbaciones asmáticas graves documentadas, a pesar de utilizar corticosteroides diarios inhalados a dosis altas, más un agonista beta2 inhalado de larga duración.
• Niños (6 a <12 años de edad): XOLAIR® está indicado para mejorar el control del asma cuando se administra como tratamiento adicional en pacientes con asma alérgica grave persistente que presentan test cutáneo positivo o reactividad in vitro a aeroalergenos perennes y síntomas frecuentes durante el día o despertares por la noche y que han presentado múltiples exacerbaciones asmáticas graves documentadas, a pesar de utilizar corticosteroides diarios inhalados a dosis altas, más un agonista beta2 inhalado de larga duración.
Urticaria espontánea crónica (UEC): XOLAIR® está indicado como tratamiento adicional, para el tratamiento de la urticaria crónica espontánea en pacientes adultos y adolescentes (a partir de 12 años) con respuesta inadecuada al tratamiento con antihistamínicos H1.
CONTRAINDICACIONES: Hipersensibilidad al principio activo o a cualquiera de los excipientes (véase Descripción y composición - Excipientes).
EMBARAZO Y LACTANCIA:
Mujeres en edad de procrear: No existen recomendaciones especiales para las mujeres en edad de procrear.
Embarazo: No se han realizado estudios comparativos adecuados con el omalizumab en mujeres embarazadas. Las IgG atraviesan la barrera placentaria. Dado que los estudios de la reproducción animal no siempre permiten predecir la respuesta humana, XOLAIR® no debe administrarse durante el embarazo, a menos que sea estrictamente necesario.
Se han estudiado los efectos del omalizumab en la reproducción de macacos. Dosis subcutáneas de hasta 75 mg de omalizumab/kg (doce veces superiores a la dosis clínica máxima) no indujeron toxicidad materna o embrionaria ni teratogenia cuando se administraron durante la organogénesis, ni tampoco efectos adversos sobre el crecimiento fetal o neonatal cuando se administraron durante el último período gravídico, el alumbramiento o la lactancia.
Aunque no se han observado efectos clínicamente significativos en los trombocitos de los pacientes, la administración de dosis de omalizumab mucho mayores que la dosis clínica se ha asociado a reducciones de los trombocitos sanguíneos de los primates estudiados, de forma dependiente de la edad, de suerte que los animales más jóvenes se vieron relativamente más afectados. En los estudios de la reproducción en macacos de Java no se apreciaron signos clínicos de trombocitopenia en las crías recién nacidas de progenitoras que habían recibido dosis de hasta 75 mg de omalizumab/kg; no obstante, no se determinó el número de trombocitos en tales crías.
Lactancia: Aunque no se ha investigado la presencia de omalizumab en la leche humana, las IgG se eliminan en ese líquido y, por consiguiente, cabe esperar que el omalizumab esté presente en la leche humana. Se desconoce la capacidad de absorción de omalizumab y de daño al lactante; se debe tener cuidado cuando se administre XOLAIR® a una madre lactante.
Se valoró la eliminación del omalizumab en la leche de las hembras de macacos que habían recibido dosis subcutáneas de 75 mg/kg/semana. Las concentraciones plasmáticas de omalizumab en las crías tras la exposición intrauterina y 28 días de lactancia variaban entre un 11% y un 94% de la concentración plasmática materna. Las concentraciones lácteas de omalizumab eran un 1,5% de la concentración sanguínea materna.
Fecundidad: No se tienen datos sobre los efectos del omalizumab en la fecundidad humana. En los estudios preclínicos especialmente diseñados para estudiar la fecundidad, como son los de apareamiento, no se observó un menoscabo de la fecundidad en los machos o hembras que recibieron dosis repetidas de hasta 75 mg de omalizumab/kg.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: La influencia de XOLAIR® sobre la capacidad para conducir y utilizar maquinas es nula o insignificante.
REACCIONES ADVERSAS:
Asma alérgica:
• Resumen del perfil toxicológico: Más de 4.400 pacientes con asma alérgica fueron randomizados durante los ensayos clínicos controlados llevados a cabo con XOLAIR®.
Las reacciones adversas notificadas con mayor frecuencia en los estudios clínicos con adultos y adolescentes (mayores de 12 años) fueron las reacciones en el lugar de la inyección, como dolor, tumefacción, eritema, prurito, así como cefaleas. En los estudios clínicos efectuados en pacientes de 6 a <12 años de edad, las reacciones adversas más frecuentes fueron las cefaleas, la fiebre y el dolor en la parte superior del abdomen. La mayoría de los acontecimientos eran de naturaleza leve o moderada.
• Resumen tabulado de las reacciones adversas observadas en los estudios clínicos: La Tabla 4 recoge las reacciones adversas registradas en los estudios clínicos en la población total con asma alérgica tratada con XOLAIR® del análisis de la seguridad, desglosadas por clase de órgano, aparato o sistema y por frecuencia. Según su frecuencia se clasifican en: Muy frecuentes (≥1/10), frecuentes (>1/100; <1/10), infrecuentes (>1/1000; <1/100), raras (<1/1000).
|
Tabla 4. Reacciones adversas observadas en los estudios clínicos |
|
|
Infecciones e infestaciones |
|
|
Infrecuente |
Faringitis |
|
Raro |
Parasitosis |
|
Trastornos del sistema inmunitario |
|
|
Raro |
Reacciones anafilácticas y otros trastornos alérgicos, producción de anticuerpos antiterapéuticos |
|
Trastornos del sistema nervioso |
|
|
Frecuente |
Cefalea** |
|
Infrecuente |
Mareos, somnolencia, parestesias, síncope |
|
Trastornos vasculares |
|
|
Infrecuente |
Hipotensión postural, crisis vasomotoras |
|
Trastornos respiratorios, torácicos y del mediastino |
|
|
Infrecuente |
Tos, broncoespasmo alérgico |
|
Raro |
Edema laríngeo |
|
Trastornos gastrointestinales |
|
|
Frecuente |
Dolor en la parte superior del abdomen* |
|
Infrecuente |
Náuseas, diarrea, signos y síntomas dispépticos |
|
Trastornos de la piel y del tejido subcutáneo |
|
|
Infrecuente |
Urticaria, exantema, prurito, fotosensibilidad. |
|
Raro |
Angioedema |
|
Trastornos generales y en el lugar de la administración |
|
|
Muy frecuente |
Fiebre (pirexia)* |
|
Frecuente |
Reacciones en el lugar de la inyección, como dolor, eritema, prurito e hinchazón. |
|
Infrecuente |
Aumento de peso, fatiga, brazos edematosos, síndrome seudogripal |
|
*: En niños de entre 6 y 12 años de edad **: Muy frecuente en niños de entre 6 y 12 años de edad |
|
Las frecuencias de las reacciones adversas observadas en los pacientes que recibieron el tratamiento activo fueron muy similares a las del grupo de comparación.
• Lista de reacciones adversas mencionadas en notificaciones espontáneas desde la comercialización del producto: Las reacciones que se describen a continuación se han identificado a través de notificaciones espontáneas.
— Trastornos del sistema inmunitario (véase Advertencias y precauciones): Reacciones anafilácticas y anafilactoides después de la primera administración o de administraciones subsiguientes.
No conocidas: Enfermedad del suero, puede incluir fiebre y linfoadenopatía.
— Trastornos de la piel y del tejido subcutáneo: No conocida: Alopecia.
— Trastornos de la sangre y del sistema linfático: No conocida: Trombocitopenia idiopática grave.
— Trastornos respiratorios, torácicos y del mediastino: No conocida: Vasculitis granulomatosa alérgica (síndrome de Churg-Strauss).
— Trastornos del aparato locomotor y del tejido conjuntivo: No conocida: Artralgia, mialgia, hinchazón de articulaciones.
Urticaria espontánea crónica (UEC):
• Resumen del perfil toxicológico: La seguridad y la tolerabilidad del omalizumab se evaluaron en 975 pacientes con UEC, de los cuales 733 recibieron omalizumab durante 12 semanas en dosis de 75, 150 o 300 mg cada cuatro semanas, y 242, el placebo. En 490 de los pacientes tratados con omalizumab la administración duró 24 semanas. De los pacientes tratados con omalizumab durante 12 semanas, 175 recibieron la dosis recomendada de 150 mg, y 412, la de 300 mg. De los pacientes tratados con omalizumab durante 24 semanas, 87 recibieron la dosis recomendada de 150 mg, y 333, la de 300 mg.
• Resumen tabulado de las reacciones adversas observadas en los estudios clínicos con las dosis recomendadas (150 o 300 mg): La Tabla 5 presenta, ordenadas por clase de órgano, aparato o sistema del MedDRA, las reacciones adversas descritas con las dosis recomendadas (150 mg y 300 mg) en los tres estudios de fase III. Tales reacciones se observaron en ≥1% de los pacientes de cualquier grupo de tratamiento, con mayor frecuencia ( ≥2%) en los que recibieron el omalizumab que en los tratados con el placebo, según una evaluación médica. Dentro de cada clase de órgano, aparato o sistema las reacciones adversas figuran por orden decreciente de frecuencia. La categoría de frecuencia de cada reacción adversa se define según la siguiente convención (CIOMS III): muy frecuente (≥1/10); frecuente (≥1/100 a <1/10); infrecuente (≥1/1000 a <1/100); rara (≥1/10 000 a < 1/1000); muy rara (<1/10 000); (de frecuencia) desconocida (no se puede calcular con los datos disponibles).
|
Tabla 5. Reacciones adversas registradas en la base de datos conjuntos de seguridad en la UEC (desde el día 1 hasta la semana 12) con las dosis recomendadas |
||||
|
Reacciones adversas |
Omalizumab – estudios Q4881g, Q4882g y Q4883g en conjunto |
Categoría de frecuencia |
||
|
Placebo N=242 |
150 mg N=175 |
300 mg N=412 |
||
|
Infecciones e infestaciones |
||||
|
Rinofaringitis |
17 (7,0%) |
16 |
27 (6,6%) |
Frecuente |
|
Sinusitis |
5 (2,1%) |
2 (1,1%) |
20 (4,9%) |
Frecuente |
|
Infección viral de las vías respiratorias altas |
0 |
4 (2,3%) |
2 (0,5%) |
Frecuente |
|
Trastornos del sistema nervioso |
||||
|
Cefalea |
7 (2,9%) |
21 (12,0%) |
25 (6,1%) |
Muy frecuente |
|
Trastornos del aparato locomotor y del tejido conjuntivo |
||||
|
Artralgia |
1 (0,4%) |
5 (2,9%) |
12 (2,9%) |
Frecuente |
Acontecimientos descritos adicionalmente en cualquier momento del período de tratamiento entre el día 1 y la semana 24 (estudios Q4881g y Q4883g) que cumplían los criterios de reacciones adversas:
• Infecciones e infestaciones: Infección de las vías respiratorias altas (placebo: 3,1%, 150 mg: 3,4%, 300 mg: 5,7%), infección de las vías urinarias (placebo: 1,8%, 150 mg: 4,6%, 300 mg: 2,4%).
• Trastornos del sistema nervioso: Cefalea sinusal [por congestión de los senos paranasales] (placebo: 0%, 150 mg: 2,3%, 300 mg: 0,3%).
• Trastornos del aparato locomotor y del tejido conjuntivo: Mialgia (placebo: 0%, 150 mg: 2,3%, 300 mg: 0,9%), dolor en las extremidades (placebo: 0%, 150 mg: 3,4%, 300 mg: 0,9%), dolor osteomuscular (placebo: 0%, 150 mg: 2,3%, 300 mg: 0,9%).
• Trastornos generales y en el lugar de la administración: Pirexia (placebo: 1,2%, 150 mg: 3,4%, 300 mg: 0,9%).
• Reacciones en el lugar de la inyección: Durante los estudios, las reacciones en el lugar de la inyección fueron más frecuentes en los pacientes del grupo del omalizumab que en los del placebo (300 mg: 2,7%, 150 mg: 0,6%, placebo: 0,8%) y consistían en inflamación, eritema, dolor, equimosis, prurito, hemorragia y urticaria.
Descripción de los aspectos toxicológicos más importantes para las indicaciones de asma alérgica y UEC: Los estudios clínicos en la UEC no han arrojado ningún dato importante que pudiera exigir una modificación de los apartados siguientes.
• Trastornos del sistema inmunológico: Ver Advertencias y precauciones.
• Experiencia en ensayos clínicos.
— Eventos cardiovasculares y cerebrovasculares de los ensayos clínicos en pacientes con asma: Se llevó a cabo un estudio de cohortes observacional de 5 años, en pacientes ≥ 12 años de edad, con asma persistente de moderada a grave y con una reacción positiva a la prueba cutánea para aeroalergenos perennes; para evaluar la seguridad a largo plazo de omalizumab, incluyendo el riesgo de neoplasia maligna.
Un total de 5007 pacientes tratados con omalizumab y 2829 no tratados con omalizumab participaron en el estudio. El porcentaje de pacientes fumadores (5 por ciento) o ex fumadores (29 por ciento) en ambos grupos fue similar. Los pacientes tenían una edad promedio de 45 años, y fueron observados por 3,7 años en promedio. Se diagnosticaron más pacientes con asma grave (50%) en el grupo de omalizumab, en comparación del grupo de los no tratados con omalizumab (23%), y 44% de los pacientes abandonaron el estudio antes del tiempo. Además el 88% de los pacientes en la cohorte tratada con omalizumab habían sido expuestos previamente a omalizumab durante un promedio de 8 meses.
Se observó una mayor tasa de incidencia (por 1000 pacientes – año) de eventos adversos graves cardiovasculares y cerebrovasculares, en pacientes tratados con omalizumab (13.4) En comparación con los pacientes no tratados con omalizumab (8.1). Entre los cuales se observaron:
|
Eventos adversos |
Omalizumab (1000 pacientes – año) |
Control (1000 pacientes – año) |
|
Ataque isquémico transitorio |
0.7 |
0.1 |
|
Infarto de miocardio |
2.1 |
0.8 |
|
Hipertensión pulmonar |
0.5 |
0 |
|
Embolia pulmonar / Trombosis venosa |
3.2 |
1.5 |
|
Angina inestable |
2.2 |
1.4 |
Asimismo, las tasas observadas para accidente cerebrovascular isquémico y muerte cardiovascular fueron similares entre ambos grupos de estudio. Los resultados sugieren un posible aumento del riesgo de eventos cardiovasculares y cerebrovasculares graves en pacientes tratados con omalizumab.
Sin embargo, el diseño del estudio observacional, la inclusión de los pacientes previamente expuestos a XOLAIR® (88%), diferencias iniciales en los factores de riesgo cardiovascular entre los grupos de tratamiento, la incapacidad de ajustar por factores de riesgo medidos, y la alta tasa de abandono del estudio limita la capacidad de cuantificar la magnitud del riesgo.
Se realizó un análisis combinado de 25 ensayos aleatorios, doble ciego, controlados con placebo, de 8 a 52 semanas de duración; para evaluar de una mejor manera el desequilibrio de eventos adversos graves a nivel cardiovascular y cerebrovascular, señalando anteriormente en el estudio de cohorte observacional. Se incluyeron un total de 3342 pacientes tratados con omalizumab y 2895 pacientes tratados con placebo en este análisis combinado. Los pacientes tenían una edad promedio de 38 años y fueron seguidos durante 6.8 meses en promedio. No se observaron desequilibrios notables en las tasas de eventos adversos graves cardiovasculares y cerebrovasculares mencionados anteriormente. Sin embargo, los resultados del análisis conjunto estuvieron basados en un bajo número de eventos, los pacientes más jóvenes, y una corta duración de seguimiento en comparación con el estudio de cohorte observacional; por lo tanto, los resultados son insuficientes para confirmar o rechazar los resultados observados en el estudio observacional de cohortes.
Plaquetas: En los estudios clínicos, pocos pacientes presentaron cifras de trombocitos por debajo del límite inferior del intervalo normal de valores de laboratorio. Ninguna de estas variaciones se asoció a episodios hemorrágicos o a una disminución de la hemoglobina. No se han notificado en seres humanos (pacientes mayores de 6 años de edad) disminuciones persistentes de las cifras trombocíticas como las que se observaron en otros primates aunque se han notificado casos aislados de trombocitopenia idiopática, incluyendo casos graves, en la fase de postcomercialización (véase Datos sobre toxicidad preclínica).
Parasitosis: En pacientes alérgicos con riesgo crónico elevado de helmintosis, un ensayo comparativo con placebo reveló un ligero aumento cuantitativo de la tasa de infestación con el omalizumab, que no fue estadísticamente significativo. El curso, la gravedad y la respuesta al tratamiento de las infestaciones permanecieron inalterados (véase Advertencias y precauciones).
INCOMPATIBILIDADES:
Vial de polvo y ampolla de disolvente para solución inyectable: XOLAIR® no debe mezclarse con ningún otro medicamento o diluyente que no sea agua estéril para inyectables.
INTERACCIONES: Las enzimas del citocromo P450, las bombas de expulsión y los mecanismos de unión a proteínas no contribuyen a la depuración de omalizumab, de modo que la probabilidad de que ocurran interacciones farmacológicas es reducida. No se han realizado estudios formales de interacción de XOLAIR® con medicamentos o vacunas.
No hay motivos farmacológicos para esperar interacciones entre los medicamentos comúnmente prescritos contra el asma o la UEC y el omalizumab.
Asma alérgica: En los estudios clínicos, XOLAIR® se utilizó normalmente asociado a corticoesteroides inhalados y orales, agonistas ß2 inhalados de acción breve o prolongada, modificadores de leucotrienos, teofilinas y antihistamínicos orales. No hubo indicios de que esos medicamentos usuales contra el asma afectasen la inocuidad de XOLAIR®. Se dispone de escasos datos sobre el uso combinado de XOLAIR® y una inmunoterapia específica (terapia de hiposensibilización). En un ensayo clínico donde XOLAIR® se administró conjuntamente con inmunoterapia, se observó que la seguridad y eficacia de XOLAIR® en combinación con inmunoterapia específica, no fue diferente a la de XOLAIR® solo.
XOLAIR® puede reducir indirectamente la eficacia de medicamentos utilizados para el tratamiento de infecciones helmínticas o por otros parásitos.
Urticaria espontánea crónica (UEC): En los estudios clínicos de la UEC, XOLAIR® se administró junto con antihistamínicos (anti-H1 o anti-H2) y antagonistas de los receptores de leucotrienos (ARLTs). No hubo indicios de que el perfil toxicológico del omalizumab administrado con esos medicamentos fuera distinto del que se le conoce en el asma alérgica. Además, un análisis farmacocinético poblacional no reveló ningún efecto importante de los antihistamínicos H2 y los ARLTs en la farmacocinética del omalizumab (véase Farmacología clínica).
Población pediátrica: Los ensayos clínicos en UCE incluyeron algunos pacientes de 12 a 17 años de edad que usaron XOLAIR® junto con antihistamínicos (anti-H1, anti-H2) y ARLTs. No se realizaron ensayos en niños menores de 12 años.
ESTUDIOS CLÍNICOS:
Asma alérgica:
• Adultos y adolescentes >12 años de edad: La seguridad y la eficacia de XOLAIR® se evaluaron en cinco ensayos comparativos con placebo, aleatorizados, con doble enmascaramiento y multicéntricos.
Dos estudios idénticos de 16 semanas (los estudios 1 y 2) demostraron la seguridad y la eficacia del omalizumab como tratamiento complementario en 1071 asmáticos alérgicos, que eran sintomáticos pese a su tratamiento con corticoesteroides inhalados (dipropionato de beclometasona de 500 µg a 1200 µg/día).
En ambos ensayos, el omalizumab fue superior al placebo en lo que concierne al criterio principal de agudización asmática (agravamiento del asma con necesidad de corticoesteroides sistémicos o una duplicación de la dosis de beclometasona inicial del paciente). El número de agudizaciones del asma fue significativamente inferior en el grupo de omalizumab (p=0,006 y p<0,001 en los estudios 1 y 2, respectivamente). Menos pacientes tratados con omalizumab experimentaron agudizaciones del asma (14,6% frente al 23,3%, p=0,009 en el estudio 1 y 12,8% frente al 30,5%, p<0,001 en el estudio 2).
En las fases de extensión con doble enmascaramiento de ambos estudios de hasta un año de duración, se siguió registrando una menor frecuencia de agudizaciones asmáticas en los pacientes tratados con omalizumab en comparación con el placebo.
En los estudios 1 y 2, se pudo demostrar una mejoría clínicamente importante de la calidad de vida del paciente asmático –valorada por medio del «Cuestionario de calidad de vida en pacientes con asma» de Juniper– al final de la fase principal del estudio de 28 semanas de duración en el grupo de XOLAIR®, en comparación con el placebo (diferencia con respecto al placebo p ≤ 0,001 en los estudios 1 y 2).
En el estudio 3, se demostró la seguridad y el efecto de «evitación de corticoesteroides» del omalizumab en 246 pacientes que padecían de asma alérgica aguda que necesitaban tratamiento diario con corticoesteroides inhalados en dosis altas (fluticasona ≥1000 µg/día) y en quienes se permitieron agonistas ß2 de acción prolongada. El estudio incluyó una fase estable de 16 semanas con corticoesteroides a los que se añadió la medicación de estudio, seguida por una fase de reducción de corticoesteroides de 16 semanas. La reducción porcentual de la dosis de corticoesteroides inhalados al final de la fase de tratamiento fue notoriamente mayor en los pacientes tratados con omalizumab que en los que recibieron el placebo (mediana del 60% frente al 50%, p=0,003). La proporción de pacientes tratados con omalizumab que pudieron disminuir su dosis de fluticasona a ≤500 µg/día fue del 60,3%, frente al 45,8% en el grupo del placebo (p>0,05).
En el estudio 4, se pudo demostrar la seguridad y la eficacia de omalizumab en 405 pacientes que padecían rinitis alérgica perenne y asma alérgica concurrente. Los pacientes aptos para participar en el estudio padecían tanto de asma alérgica como de rinitis alérgica perenne. Dichos pacientes recibieron omalizumab o placebo durante 28 semanas como tratamiento complementario de ≥400 µg de budesónida administrada con el dispositivo Turbohaler. Se permitió el uso de agonistas ß2 inhalados de acción prolongada (39%) y corticoesteroides nasales (17%).
En el estudio 4, otros criterios igualmente importantes de valoración fueron la incidencia de agudizaciones del asma (agravamiento del asma que necesitaba corticoesteroides sistémicos o la duplicación de la dosis de budesónida inicial del paciente) y la proporción de pacientes de cada grupo terapéutico con una mejoría ≥1,0 desde el inicio al final de la fase de tratamiento en ambas evaluaciones de la calidad de vida específicas del asma y la rinitis (Evaluación de la calidad de vida de Juniper).
Los pacientes tratados con omalizumab presentaban una incidencia significativamente menor de agudizaciones asmáticas que los pacientes del grupo del placebo (omalizumab: 20,6% frente al placebo: 30,1%, p=0,02) y hubo una proporción notoriamente mayor de pacientes tratados con omalizumab que con el placebo que mejoraron ≥1,0 puntos según ambas evaluaciones de la calidad de vida específicas del asma y la rinitis (omalizumab: 57,7% frente al placebo: 40,6%, p<0,0001).
La reducción de las agudizaciones y las mejoras de la calidad de vida de los pacientes tratados con omalizumab se observaron en el contexto de mejorías estadísticamente significativas en los síntomas del asma y la rinitis y la función pulmonar, en comparación con el placebo.
El estudio 5, de 28 semanas de duración, demostró la eficacia y la seguridad de XOLAIR® en 419 pacientes de 12 a 79 años de edad con asma alérgica grave que presentaban una reducción de la función pulmonar (volumen espiratorio máximo en el primer segundo [FEV1]: 40-80% del previsto) y un control insuficiente de los síntomas asmáticos a pesar de recibir tratamiento con >1000 µg de dipropionato de beclometasona (o su equivalente) asociado a un agonista ß2 de acción prolongada. Los pacientes admitidos en el estudio habían padecido múltiples agudizaciones del asma que exigían tratamiento con corticoesteroides sistémicos o habían sido hospitalizados o atendidos en un Servicio de urgencias a causa de una agudización asmática intensa el año anterior, pese a su tratamiento continuo con corticoesteroides inhalados en dosis elevadas y agonistas ß2 de acción prolongada. Se administró XOLAIR® o placebo por vía subcutánea como tratamiento complementario de >1000 µg (o cantidad equivalente) más un agonista ß2 de acción prolongada. Se permitió la administración de un tratamiento de mantenimiento a base de corticoesteroides orales (22%), teofilina (27%) o antileucotrienos (35%). En la fase de tratamiento no se modificó la terapia antiasmática concomitante.
El porcentaje de agudizaciones del asma que exigían tratamiento con tandas cortas de corticoesteroides sistémicos fue el criterio principal de valoración. El omalizumab redujo el porcentaje de agudizaciones asmáticas en un 19% (p=0,153). Otras evaluaciones, que no arrojaron ninguna significación estadística (p<0,05) a favor de XOLAIR®, revelaron reducciones en agudizaciones intensas (en las que la función pulmonar del paciente se redujo a menos del 60% de la cifra óptima individual, lo cual necesitó corticoesteroides sistémicos) y en consultas de urgencia relacionadas con el asma (entre ellas hubo consultas médicas no programadas, consultas al Servicio de urgencias y hospitalizaciones), así como mejoras en la evaluación general del médico de la eficacia práctica del tratamiento, la calidad de vida relacionada con el asma (CVA), los síntomas asmáticos y la función pulmonar. La evaluación general del médico se llevó a cabo en los cinco estudios mencionados anteriormente, como medida amplia de control del asma a cargo del médico terapeuta. El médico tuvo en cuenta el flujo espiratorio máximo, los síntomas diurnos y nocturnos, el uso de tratamientos de rescate, la espirometría y las agudizaciones. En los cinco estudios sin excepción una proporción significativamente mayor de pacientes tratados con XOLAIR® lograron una marcada mejoría o un control completo del asma en comparación con los pacientes que utilizaron el placebo.
• Niños 6 a <12 años de edad: El principal aval de la seguridad y la eficacia de XOLAIR® en el grupo de 6 a <12 años de edad proviene de un ensayo comparativo con placebo, aleatorizado, de doble enmascaramiento y multicéntrico (el estudio 6) y de un estudio complementario (estudio 7).
El estudio 6 fue un ensayo de 52 semanas de duración en el que se evaluó la seguridad y la eficacia de XOLAIR® como tratamiento complementario en 628 niños con asma alérgica mal controlada a pesar de su tratamiento con corticoesteroides inhalados convencionales (≥ 200 µg/día de fluticasona administrada con un inhalador de polvo seco o su equivalente) con o sin otros tratamientos antiasmáticos. Los pacientes seleccionados para participar en el estudio padecían asma desde hacía más de un año, tenían resultados positivos de al menos un aeroalérgeno perenne en las pruebas alérgicas cutáneas y presentaban antecedentes de manifestaciones clínicas de asma persistente moderada o grave (diurnas o nocturnas), así como antecedentes de agudizaciones en el año previo a su admisión en el estudio. Se permitió el uso de agonistas ß2 de acción prolongada (67,4%), antileucotrienos (36,6%) y corticoesteroides orales (1,3%) como tratamiento de fondo. Durante las 24 primeras semanas de tratamiento se mantuvieron constantes las dosis iniciales de corticoesteroides de cada paciente y durante las 28 semanas siguientes se permitió un ajuste de los corticoesteroides inhalados.
Una agudización clínicamente significativa se definió como un agravamiento de los síntomas del asma, según el criterio clínico del investigador, que requiere una duplicación de la dosis inicial de corticoesteroides inhalados durante al menos 3 días o un tratamiento de rescate con corticoesteroides sistémicos (orales o intravenosos) durante al menos 3 días.
La tasa de agudizaciones durante el periodo de tratamiento con doble enmascaramiento de 52 semanas en el grupo de pacientes tratados con XOLAIR® que presentaban un FEV1 > 80% al inicio del estudio disminuyó un 43% con respecto al grupo que recibió el placebo (p<0,001). En comparación con los pacientes que recibieron el placebo, los pacientes tratados con XOLAIR® presentaron una reducción estadísticamente significativa de la tasa de agudizaciones, independientemente del uso concomitante de agonistas ß2 de acción prolongada al inicio del estudio. Esta reducción fue del 45% en los pacientes que recurrieron a agonistas ß2 de acción prolongada y del 42% en los pacientes que no los utilizaron (p<0,001 y p=0,011, respectivamente).
En el estudio 7, de 28 semanas de duración, comparativo, con doble enmascaramiento, se evaluó principalmente la seguridad en 334 pacientes suficientemente controlados con corticoesteroides inhalados. Las dosis iniciales de corticoesteroides se mantuvieron constantes durante las primeras 16 semanas, y durante las 12 semanas siguientes se redujeron. En el estudio se evaluó la reducción porcentual de la dosis de dipropionato de beclometasona y la proporción de pacientes que consiguieron tal reducción a las 28 semanas. La reducción porcentual de la dosis de dipropionato de beclometasona a las 28 semanas fue mayor en el grupo tratado con XOLAIR® que en el grupo del placebo (mediana de reducciones: 100% frente al 66,7%; p=0,001), así como la proporción de pacientes que pudieron reducir la dosis de dipropionato de beclometasona (p=0,002). La frecuencia e incidencia de episodios de agudización asmática durante la fase de reducción de la dosis de corticoesteroide también fue menor en el grupo del omalizumab (tasa media de 0,42 frente a 0,72, p< 0,001; porcentaje de pacientes con agudizaciones del 18% frente al 39%, p<0,001). Durante las primeras 16 semanas del periodo terapéutico de 24 semanas se hizo patente la superioridad del omalizumab con respecto a la disminución de la incidencia y frecuencia de agudizaciones. En el 55,7% de los pacientes tratados con el omalizumab hubo una reducción completa (100%) de la dosis de corticoesteroide al final del periodo de tratamiento de 28 semanas, en comparación con el 43,2% de los pacientes que recibieron el placebo. Además, en más pacientes del grupo del omalizumab hubo una reducción ≥50% en la dosis de corticoesteroide en comparación con el placebo (80,4% frente al 69,5%, p=0,017).
Los dos estudios mencionados anteriormente (6 y 7) incluyeron una evaluación general realizada por el médico terapeuta como medida amplia del control del asma. El médico tuvo en cuenta el flujo espiratorio máximo, los síntomas diurnos y nocturnos, el uso de tratamientos de rescate, la espirometría y las agudizaciones. En ambos estudios, la proporción de pacientes que presentaron una mejoría importante o un control completo del asma fue significativamente mayor en los pacientes tratados con XOLAIR® que en los que recibieron el placebo.
Urticaria espontánea crónica (UEC): El programa de desarrollo clínico de fase III en la UEC comprendió tres estudios comparativos con placebo, con grupos paralelos, aleatorizados, con doble enmascaramiento y multicéntricos, los estudios Q4881g, Q4882g y Q4883g.
En los estudios Q4881g y Q4882g, se evaluaron la eficacia y la seguridad de la administración de 75, 150 o 300 mg de XOLAIR® cada 4 semanas durante 24 y 12 semanas, respectivamente, con un período de observación sin tratamiento de 16 semanas, en pacientes (12–75 años) con UEC resistente pese a su tratamiento con antihistamínicos H1.
En el estudio Q4883g, se evaluaron la seguridad y la eficacia de la administración de 300 mg de XOLAIR® cada 4 semanas durante 24 semanas, con un período de observación sin tratamiento de 16 semanas, en pacientes (12–75 años) con UEC resistente pese a su tratamiento con antihistamínicos H1 o H2 o con ARL.
|
Tabla 6. Criterios de eficacia |
|
|
Variación de la PIP semanal con respecto al inicio al cabo de 12 semanas (PIP: puntuación de la intensidad del prurito; escala de 0–21) |
Criterio principal en los estudios Q4881g y Q4882g Criterio secundario en el estudio Q4883g (en elque se evaluó principalmente la seguridad) |
|
Tiempo transcurrido hasta observar una DMIa (disminución ≥5 puntos con respecto al inicio) en la PIP semanal durante el período de 12 semanas |
Criterios secundarios en los tres estudios Q4881g, Q4882g y Q4883g
|
|
Variación de la puntuación de la actividad urticarial durante un período de 7 días con respecto al inicio al cabo de 12 semanas (PAU7b; escala de 0-42) |
|
|
Proporción de pacientes con una puntuación de la actividad urticarial ≤ 6 durante un período de 7 días al cabo de 12 semanas (PAU7 b ≤ 6) |
|
|
Proporción de pacientes con una puntuación de la actividad urticarial = 0 durante un período de 7 días al cabo de 12 semanasc (PAU7b = 0) |
|
|
Variación de la puntuación semanal del número de ronchas con respecto al inicio al cabo de 12 semanas |
|
|
Variación del ICVD (índice de calidad de vida en dermatología) general con respecto al inicio al cabo de 12 semanas |
|
|
Proporción de pacientes con días transcurridos sin angioedema desde la semana 4 hasta la semana 12d |
|
|
a DMI: Diferencia mínimamente importante b PAU7: Criterio compuesto de la intensidad del prurito y del número de ronchas, evaluados a diario y totalizados semanalmente. c Análisis a posteriori del estudio Q4882g d La proporción (media) de días transcurridos sin angioedema desde la semana 4 hasta la semana 12 se calculó considerando toda la población del estudio, que incluye a los pacientes sin síntomas de angioedema. |
|
En los estudios Q4881g y Q4882g, la dosis de 75 mg no siempre permitió satisfacer el criterio principal de eficacia (variación de la PIP semanal con respecto al inicio al cabo de 12 semanas) u otros criterios secundarios. Se estimó que no era eficaz y por eso mismo no se abordará más aquí.
Variación de la PIP semanal con respecto al inicio al cabo de 12 semanas: Ambas dosis de 150 y 300 mg satisficieron el criterio de eficacia principal (variación de la PIP semanal con respecto al inicio al cabo de 12 semanas ) en los estudios Q4881g y Q4882g, y lo mismo sucedió con la dosis de 300 mg en el estudio Q4883g (véase la Tabla 7).
|
Tabla 7. Variación de la PIP semanal con respecto al inicio al cabo de 12 semanas, estudios Q4881g, Q4882g y Q4883g (Población IDTm*) |
|||
|
Placebo |
Omalizumab 150 mg |
Omalizumab 300 mg |
|
|
Estudio Q4881g |
|||
|
N |
80 |
80 |
81 |
|
Media (± DE) |
-3,63 (5,22) |
-6,66 (6,28) |
-9,40 (5,73) |
|
Diferencia de medias mínimo cuadráticas entre el omalizumab y el placebo1 |
– |
-2,95 |
-5,80 |
|
IC (del 95%) de la diferencia |
– |
-4,72,-1,18 |
-7,49,-4,10 |
|
Valor de p de la comparación con el placebo2 |
– |
0,0012 |
<0,0001 |
|
Estudio Q4882g |
|||
|
N |
79 |
82 |
79 |
|
Media (± DE) |
-5,14 (5,58) |
-8,14 (6,44) |
-9,77 (5,95) |
|
Diferencia de medias mínimo cuadráticas entre el omalizumab y el placebo1 |
– |
-3,04 |
-4,81 |
|
IC (del 95%) de la diferencia |
– |
-4,85, -1,24 |
-6,49,-3,13 |
|
Valor de p de la comparación con el placebo2 |
– |
0,0011 |
<0,0001 |
|
Estudio Q4883g |
|||
|
n |
83 |
– |
252 |
|
Media (± DE) |
-4,01 (5,87) |
– |
-8,55 (6,01) |
|
Diferencia de medias mínimo cuadráticas entre el omalizumab y el placebo1 |
– |
– |
-4,52 |
|
IC (del 95%) de la diferencia |
– |
– |
-5,97, -3,08 |
|
Valor de p de la comparación con el placebo2 |
– |
– |
<0,0001 |
|
*Población de análisis por intención de tratar modificada (o población IDTm): Incluye a todos los pacientes que fueron aleatorizados y que recibieron por lo menos una dosis del medicamento de estudio. Se imputó la BOCF (observación realizada al inicio) cuando no se disponía de datos. 1 La media mínimo cuadrática se estimó usando un modelo de análisis de la covarianza (ANCOVA). Los estratos fueron la puntuación semanal de la intensidad del prurito al inicio del estudio (< 13 frente a ≥13) y el peso al inicio del estudio (< 80 kg frente a ≥ 80 kg). 2 El valor de p deriva de la prueba de la t del modelo de ANCOVA |
|||
La Figura 1 presenta la puntuación semanal media de la intensidad del prurito obtenida a lo largo del estudio Q4881g. Las puntuaciones semanales medias de la intensidad del prurito disminuyeron considerablemente en ambos grupos terapéuticos; el efecto máximo se registró alrededor de la semana 12 y fue constante durante el período de tratamiento de 24 semanas. Los estudios Q4883g (administración de 300 mg durante el período de tratamiento de 24 semanas) y Q4882g (administración de 150 o 300 mg durante el período de tratamiento de 12 semanas) arrojaron resultados similares a los del estudio Q4881g.
En los tres estudios (véase la Figura 1 correspondiente al estudio Q4881g), la puntuación semanal media de la intensidad del prurito aumentó gradualmente con ambas dosis de omalizumab durante el período de observación sin tratamiento de 16 semanas, lo cual es indicativo de una recidiva de los síntomas. Los valores medios al final del período de observación fueron similares a los del grupo del placebo, pero menores que los respectivos valores medios iniciales.
Figura 1. Puntuación semanal media de la intensidad del prurito en función del tiempo, estudio Q4881g (BOCF, población IDTm)
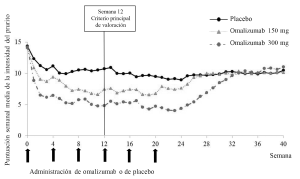
BOCF= (imputación de la) observación realizada al inicio; IDTm= población por intención de tratar modificada
Tiempo transcurrido hasta observar una DMI en la PIP semanal durante el período de 12 semanas: En los estudios Q4881g y Q4882g, el tiempo mediano transcurrido hasta observar una DMI de 5 puntos en la puntuación semanal de la intensidad del prurito fue de 2 semanas en el grupo de 150 mg (p=0,0301 en el estudio Q4881g; p=0,0101 en el estudio Q4882g), de 1 semana (p<0,0001) en el grupo de 300 mg y de 4 semanas con el placebo. Parecidos resultados se obtuvieron en el estudio Q4883g, en el que la DMI se observó al cabo de una mediana de 2 semanas en el grupo de 300 mg (p<0,0001) o de 5 semanas en el grupo del placebo.
Variación de la PAU7 con respecto al inicio al cabo de 12 semanas: En los estudios de fase III, se apreció una diferencia estadísticamente significativa entre los grupos tratados con omalizumab (150 o 300 mg) y el grupo del placebo en la variación media con respecto al inicio de la PAU7 al cabo de 12 semanas (Figura 2 del estudio Q4881g). Se obtuvieron resultados estadísticamente significativos (p<0,0001) en el grupo de 300 mg de los tres estudios y en el grupo de 15 mg de los estudios Q4881g (p=0,0008) y Q4882g (p=0,0001).
La Figura 2 muestra la PAU7 media a lo largo del estudio Q4881g y revela una disminución significativa con respecto a los valores iniciales en ambos grupos terapéuticos, con un efecto máximo alrededor de la semana 12. La magnitud del efecto se mantuvo durante el período de tratamiento de 24 semanas. En los estudios Q4883g (300 mg administrados durante 12 semanas) y Q4882g (150 o 300 mg administrados durante 24 semanas) se obtuvieron resultados similares a los del estudio Q4881g.
En los tres estudios (véase la Figura 2 correspondiente al estudio Q4881g), la PAU7 aumentó gradualmente en ambos grupos del omalizumab durante el período de observación sin tratamiento de 16 semanas, lo cual era indicativo de una recidiva de los síntomas. Los valores medios al final del período de observación fueron similares a los del grupo del placebo, pero menores que los respectivos valores medios iniciales.
Figura 2. PAU7 media en función del tiempo, estudio Q4881g (BOCF, población IDTm)
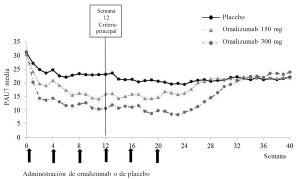
BOCF=(imputación de la) observación realizada al inicio; IDTm= población por intención de tratar modificada; PAU7=puntuación de la actividad urticarial durante un período de 7 días
Proporción de pacientes con una PAU7 ≤ 6 al cabo de 12 semanas: Los porcentajes de respuesta (UAS7 ≤6) al cabo de 12 semanas fueron todos estadísticamente significativos, de 52–66% en el grupo de 300 mg (51,9% en el estudio Q4881g, 65,8% en el estudio Q4882g y 52,4% en el estudio Q4883g; p<0,0001) o de 40–43% en el grupo de 150 mg (40,0% en el estudio Q4881g, 42,7% en el estudio Q4882g; p<0,001), en comparación con 11–19% del grupo del placebo (11,3% en el estudio Q4881g, 19,0% en el estudio Q4882g y 12,0% en el estudio Q4883g).
Proporción de pacientes con una PAU = 0 al cabo de 12 semanas: La proporción de pacientes que habían respondido por completo al tratamiento (PAU7 = 0) fue estadísticamente significativa en el grupo de 300 mg al cabo de 12 semanas –a saber, de 34–44% (35,8% en el estudio Q4881g, 44,3% en el estudio Q4882g y 33,7% en el estudio Q4883g, todos con una p<0,0001)– y mayor que la observada en el grupo de 150 mg, que fue de 15,0% en el estudio Q4881g y de 22,0% en el estudio Q4882g, en comparación con 5-9% del grupo del placebo (8,8% en el estudio Q4881g, 5,1% en el estudio Q4882g y 4,8% en el estudio Q4883g).
Variación de la puntuación semanal del número de ronchas con respecto al inicio al cabo de 12 semanas: En los tres estudios de fase III, la variación media de la puntuación semanal del número de ronchas con respecto al inicio al cabo de 12 semanas en los grupos de tratamiento con 300 mg fue estadísticamente significativa (p<0,001); todos ellos presentaban un descenso de la puntuación del número de ronchas en comparación con el grupo del placebo (-11,35 en el estudio Q4881g, -11,97 en el estudio Q4882g y -10,46 en el estudio Q4883g y de -4,37, -5,22 y -4,49 en los respectivos grupos del placebo). En los grupos de tratamiento con 150 mg, la variación media fue de -7,78 (p=0,0017) en el estudio Q4881g y de -9,75 (p<0,001) en el estudio Q4882g.
Proporción de días transcurridos sin angioedema desde la semana 4 hasta la semana 12: En los tres estudios de fase III, desde la semana 4 hasta la semana 12 de tratamiento, los pacientes del grupo de 300 mg gozaron sistemáticamente de la mayor proporción media de días transcurridos sin angioedema (96,1% en el estudio Q4881g; 95,5% en el estudio Q4882g; 91% en el estudio Q4883g; todos los valores p<0,001) en comparación con el grupo del placebo (88,2%, 89,2%, 88,1%, respectivamente). En los grupos de tratamiento con 150 mg, durante el mismo período, la proporción media de días transcurridos sin angioedema en los estudios Q4881g y Q4882g fue de 89,6% y 91,6% respectivamente, y no se apreció ninguna diferencia estadísticamente significativa con respecto al placebo.
Variación del ICVD (índice de calidad de vida en dermatología) general con respecto al inicio al cabo de 12 semanas: En los tres estudios de fase III, la variación media con respecto al inicio del ICVD general en los grupos de tratamiento con 300 mg fue mayor que la observada en el grupo del placebo, con una diferencia estadísticamente significativa (p<0,001) entre ambos grupos. En el estudio Q4881g se apreció una mejora de 10,3 puntos, en el Q4882g, de 10,2, y en el Q4883g, de 9,7, mientras que la mejora en los correspondientes grupos del placebo fue de 6,1, 6,1 y 5,1, respectivamente. En los grupos de tratamiento con 150 mg, la variación media fue de 8,0 puntos (p=0,2286) en el estudio Q4881g, de 8,3 puntos (p=0,0215) en el estudio Q4882g y de 6,1 en cada uno de los respectivos grupos del placebo.
Eficacia tras 24 semanas de tratamiento: La Tabla 8 recoge los resultados obtenidos al cabo de 24 semanas de tratamiento. La magnitud de la respuesta al tratamiento fue similar a la que se apreció tras 12 semanas de tratamiento.
|
Tabla 8. Resultados de eficacia al cabo de 24 semanas de tratamiento, estudios Q4881g y Q4883g (población IDTm*) |
||||
|
Parámetro Estudio |
Semana |
Placebo |
Omalizumab 150 mg |
Omalizumab 300 mg |
|
Variación de la PIP semanal con respecto al inicio (BOCF), media de valores |
||||
|
Estudio Q4881g |
Semana 24 |
-5,41 |
-6,47 |
-9,84** |
|
Estudio Q4883g |
Semana 24 |
-4,03 |
NP |
-8,60** |
|
Variación de la PAU7 con respecto al inicio (BOCF), media de valores |
||||
|
Estudio Q4881g |
Semana 24 |
-11,73 |
-14,21 |
-22,11** |
|
Estudio Q4883g |
Semana 24 |
-8,85 |
NP |
-19,15** |
|
Proporción (%) de pacientes con una PAU7 ≤ 6 |
||||
|
Estudio Q4881g |
Semana 24 |
25,0 |
36,3 |
61,7** |
|
Estudio Q4883g |
Semana 24 |
16,9 |
NP |
55,6** |
|
Proporción (%) de pacientes con una PAU7 = 0 |
||||
|
Estudio Q4881g |
Semana 24 |
12,5 |
20,0 |
48,1** |
|
Estudio Q4883g |
Semana 24 |
3,6 |
NP |
42,5** |
|
*Población de análisis por intención de tratar modificada (o población IDTm): Incluye a todos los pacientes que fueron aleatorizados y que recibieron por lo menos una dosis del medicamento de estudio ** Valor de p≤0,0001 para el correspondiente estadístico del análisis de comparación del tratamiento administrado y el placebo NP: No procede. BOCF: (Imputación de la) observación realizada al inicio. |
||||
DATOS SOBRE TOXICIDAD PRECLÍNICA:
No se han observado signos de reacciones anafilácticas sistémicas por desgranulación de los mastocitos en macacos adultos o jóvenes. En todos los estudios con primates se hallaron complejos circulantes de omalizumab:IgE, pero tras la administración de omalizumab no se encontraron signos de enfermedad mediada por inmunocomplejos en órgano alguno (incluido el riñón). Los complejos de omalizumab:IgE no fijan complemento, ni median la citotoxicidad dependiente del complemento.
La administración repetida de dosis de hasta 250 mg de omalizumab/kg (más de 14 veces superiores a la dosis clínica máxima admisible de 17,5 mg/kg) fue bien tolerada en primates adultos y jóvenes, salvo la disminución dosis-dependiente del número de trombocitos observada en algunas especies con concentraciones séricas generalmente superiores a la exposición humana máxima que se utilizó en los estudios clínicos fundamentales. Los monos jóvenes fueron más sensibles que los adultos a estos efectos trombocíticos. Además, los macacos mostraban signos de inflamación y hemorragia aguda en el lugar de la inyección, indicativos de una reacción inmunitaria local a la administración subcutánea repetida de una proteína heteróloga. No se han realizado estudios formales del poder cancerígeno del omalizumab.
Se han detectado anticuerpos antiomalizumábicos en algunos monos tras la administración subcutánea o intravenosa. Cabe esperar que ello suceda al administrar una proteína heteróloga. Algunos animales no pudieron ser evaluados debido a las concentraciones séricas elevadas de omalizumab, a las concentraciones elevadas de IgE o a ambas cosas a la vez. No obstante, los animales mantuvieron las elevadas concentraciones séricas de omalizumab durante el período de tratamiento de los estudios, y no hubo signos evidentes de toxicidad debido a la presencia de anticuerpos contra el omalizumab.
Los estudios de la función reproductora, la eliminación en la leche y la fecundidad se describen en el apartado Embarazo y lactancia.
ADVERTENCIAS Y PRECAUCIONES:
Trastornos del sistema inmunológico.
Neoplasia maligna: En los estudios clínicos realizados en adultos y adolescentes (≥ 12 años de edad) con asma y otras enfermedades alérgicas, se observaron neoplasias malignas en 20 de 4127 (0,5%) de los pacientes tratados con omalizumab, en comparación con los pacientes control que fueron 5 de 2236 (0,2%). Las neoplasias malignas observadas en pacientes tratados con omalizumab fueron de tipo: mama, piel (no melanoma), próstata, melanoma, y parótida; los que ocurrieron más de una vez; y otros cinco tipos que ocurrieron una vez cada uno. La mayoría de los pacientes se observaron durante menos de 1 año. El impacto de una exposición más prolongada a omalizumab, o su uso en pacientes con un mayor riesgo de neoplasia maligna (por ejemplo: fumadores, ancianos) no se conoce.
En un estudio observacional de 5007 pacientes tratados con omalizumab y 2829 no tratados con omalizumab, con asma moderada a grave persistente y una reacción a la prueba cutánea positiva o reactividad in vitro a aeroalergenos perennes, los pacientes fueron seguidos durante un máximo de 5 años. En este estudio, las tasas de incidencia de tumores malignos primarios (por 1000 pacientes-año) fueron similares entre los tratados con XOLAIR® (12,3) y los pacientes no tratados con omalizumab (13.0). Sin embargo, las limitaciones del estudio imposibilitan definitivamente descartar un riesgo de neoplasia maligna con omalizumab. Las limitaciones del estudio incluyen: el diseño observacional del estudio, el sesgo introducido al permitir la inscripción de los pacientes previamente expuestos a omalizumab (88%), un criterio inicial para la participación que excluía a pacientes con antecedentes de cáncer o una condición premaligna, y una alta tasa de abandono de los participantes en el estudio (44%).
Reacciones alérgicas de tipo I: Como sucede con cualquier otra proteína, pueden sobrevenir reacciones alérgicas locales o generales, incluso reacciones anafilácticas, cuando se administra XOLAIR®. Por consiguiente, se debe tener a mano algún medicamento para el tratamiento inmediato de las reacciones anafilácticas tras la administración de XOLAIR®. Se debe comunicar al paciente la posibilidad de que ocurran dichas reacciones y que, en caso de padecerlas, acuda al médico de inmediato. Rara vez se han observado reacciones anafilácticas en los ensayos clínicos (véase Reacciones adversas).
Desde la comercialización del producto se han registrado reacciones anafilácticas y anafilactoides después de la primera administración o de administraciones posteriores de XOLAIR®. Casi todas esas reacciones ocurrieron en un plazo de dos horas, pero algunas después de dos horas.
Al igual que sucede con otros anticuerpos monoclonales humanizados obtenidos por ingeniería genética, los pacientes pueden, en contadas ocasiones, generar anticuerpos contra el omalizumab (véase Reacciones adversas).
Enfermedad del suero: Se han registrado casos de enfermedad del suero y reacciones similares a la enfermedad del suero (que son reacciones de hipersensibilidad retardada de tipo III) en pacientes tratados con anticuerpos monoclonales humanizados, como el omalizumab. Estas reacciones comienzan típicamente 1–5 días después de la administración de la primera inyección o de las inyecciones siguientes e incluso después de tratamientos más prolongados. Entre los síntomas indicativos de la enfermedad del suero figuran artritis o artralgias, exantemas (urticaria u otras formas), fiebre y linfadenopatías. Los antihistamínicos y los corticoesteroides pueden ser útiles para prevenir o tratar este trastorno y hay que pedir a los pacientes que refieran cualquier síntoma sospechoso.
Síndrome de Churg-Strauss y síndrome hipereosinofílico: Los pacientes con asma grave pueden presentar raramente síndrome hipereosinofílico sistémico o vasculitis granulomatosa eosinofílica alérgica (Síndrome de Churg-Strauss), los cuales son normalmente tratados con corticosteroides sistémicos.
En raras ocasiones, los pacientes en tratamiento con medicamentos antiasmáticos, incluyendo omalizumab, pueden presentar o desarrollar eosinofilia sistémica y vasculitis. Estas reacciones están normalmente asociadas con la reducción del tratamiento con corticosteroides orales.
En estos pacientes, los médicos deberán estar alerta ante el desarrollo de eosinofilia importante, rash vasculítico, empeoramiento de los síntomas pulmonares, anormalidades en el seno paranasal, complicaciones cardíacas, y/o neuropatía.
Deberá considerarse la interrupción del tratamiento con omalizumab en todos aquellos casos graves que cursen con alteraciones del sistema inmune mencionadas anteriormente.
Parasitosis: La IgE puede estar implicada en reacciones inmunitarias a ciertas infestaciones. Un ensayo comparativo con placebo efectuado en pacientes alérgicos con riesgo crónico elevado de helmintosis reveló un leve aumento de la tasa de infestación con el omalizumab, pero el curso, la gravedad y la respuesta al tratamiento de la infestación no presentaban diferencias.
La tasa de helmintosis en el programa clínico general, que no fue diseñado para detectar tales infestaciones, fue inferior a 1 de cada 1000 pacientes. No obstante, podría ser necesario tener cautela en pacientes expuestos a un elevado riesgo de contraer helmintosis, especialmente cuando se viaje a zonas donde las helmintosis sean endémicas. Si los pacientes no responden al tratamiento antihelmítico recomendado, hay que pensar en retirar el tratamiento con XOLAIR®.
Generales: XOLAIR® no está indicado para el tratamiento de las agudizaciones del asma, de los broncoespasmos agudos ni de los estados asmáticos de carácter agudo.
XOLAIR® no ha sido estudiado en pacientes con síndrome de hiperinmunoglobulinemia E o aspergilosis broncopulmonar alérgica, ni como tratamiento preventivo de reacciones anafilácticas.
XOLAIR® no ha sido suficientemente estudiado como tratamiento de la dermatitis atópica, la rinitis alérgica o la alergia alimentaria.
Tampoco ha sido estudiado en pacientes con enfermedades autoinmunitarias, procesos mediados por inmunocomplejos o disfunción renal o hepática subyacente. Se debe tener cautela cuando se administre XOLAIR® a estos pacientes.
No se recomienda la interrupción brusca de la administración de corticoesteroides sistémicos o inhalados después de instaurar la terapia con XOLAIR®. Las reducciones de los corticoesteroides deben realizarse bajo supervisión directa de un médico y posiblemente deban hacerse de forma gradual.
Recomendación al usuario: Este producto debe administrarse únicamente por vía subcutánea. No debe administrarse por vía intravenosa ni intramuscular.
POSOLOGÍA Y ADMINISTRACIÓN: El tratamiento con XOLAIR® debe iniciarlo un médico experimentado en el diagnóstico y tratamiento del asma grave persistente o urticaria espontánea crónica.
Posología en el asma alérgica: La dosis adecuada y la frecuencia de administración de XOLAIR® se eligen con arreglo a la concentración basal de IgE (UI/ml), medida antes de iniciar el tratamiento, y al peso corporal (kg). A efectos de la asignación de la dosis, antes de la administración inicial, se debe valorar la concentración de IgE de los pacientes mediante un ensayo comercial de IgE sérica total. Según estas determinaciones, podrían necesitarse entre 75 y 600 mg de XOLAIR® repartidos en 1 a 4 inyecciones en cada administración.
Era menos probable que experimentaran beneficio los pacientes con un valor de IgE inferior a 76 UI/mL. Los médicos prescriptores deberán asegurar que los pacientes adultos y adolescentes con una IgE por debajo de 76 UI/ml y los niños (6 a < 12 años de edad) con una IgE por debajo de 20 UI/ml presenten una reactividad in vitro inequívoca (RAST) al alérgeno perenne antes de iniciar el tratamiento.
Véanse la Tabla 1 para la conversión de la dosis, y las Tablas 2 y 3 para asignar dosis a niños (mayores de 6 y menores de 12 años) y a adultos y adolescentes (mayores de 12 años). No se debe administrar XOLAIR® a pacientes cuya concentración basal de IgE o cuyo peso corporal (en kg) excedan los límites indicados en la tabla de administración.
La dosis máxima recomendada es de 600 mg de omalizumab cada dos semanas.
|
Tabla 1. Conversión de la dosis en número de viales, número de inyecciones y volumen total inyectado en cada administración |
||||
|
Dosis (mg) |
Número de viales |
Número de inyecciones |
Volumen total inyectado (ml) |
|
|
75 mg a |
150 mg b |
|||
|
75 |
1c |
0 |
1 |
0.6 |
|
150 |
0 |
1 |
1 |
1.2 |
|
225 |
1c |
1 |
2 |
1.8 |
|
300 |
0 |
2 |
2 |
2.4 |
|
375 |
1c |
2 |
3 |
3.0 |
|
450 |
0 |
3 |
3 |
3.6 |
|
525 |
1c |
3 |
4 |
4.2 |
|
600 |
0 |
4 |
4 |
4.8 |
|
a 0,6 ml = volumen máximo suministrado por vial (XOLAIR® 75 mg). b 1,2 ml = volumen máximo suministrado por vial (XOLAIR® 150 mg). c o utilizar 0,6 ml de un vial de 150 mg. |
||||
Duración del tratamiento, supervisión y ajustes posológicos: XOLAIR® está previsto como tratamiento a largo plazo. Los ensayos clínicos han demostrado que son necesarias un mínimo de 12 – 16 semanas para que el tratamiento con XOLAIR® demuestre efectividad. A las 16 semanas de iniciar el tratamiento con XOLAIR®, los pacientes deberán ser evaluados por su médico con respecto a la efectividad del tratamiento antes de administrar inyecciones posteriores. La decisión de continuar con XOLAIR® tras las 16 semanas, o en ocasiones posteriores, debe estar basada en si se observa una notable mejoría en el control global del asma.
La retirada del tratamiento suele provocar un retorno de las concentraciones elevadas de IgE libre y de los síntomas asociados. Los valores de IgE total son altos durante el tratamiento y siguen siéndolo hasta un año después de haberlo retirado. Por consiguiente, de nada sirve volver a medir las concentraciones de IgE durante el tratamiento con XOLAIR® para determinar la dosis. La determinación de la dosis después de haber interrumpido el tratamiento durante menos de un año debe basarse en las concentraciones séricas de IgE obtenidas al determinar la dosis inicial. Si el tratamiento con XOLAIR® se ha interrumpido durante un año o más, se pueden volver a medir las concentraciones séricas de IgE total para determinar la dosis.
Las variaciones significativas del peso corporal exigen una adaptación posológica (véanse las Tablas 2 y 3).
|
Tabla 2. Administración cada 4 semanas. Dosis de XOLAIR® (mg por dosis) administrada por inyección subcutánea cada 4 semanas |
||||||||||
|
IgE basal (UI/ml) |
Peso corporal (kg) |
|||||||||
|
≥20-25 |
>25-30 |
>30–40 |
>40–50 |
>50–60 |
>60–70 |
>70–80 |
>80–90 |
>90– 125 |
>125–150 |
|
|
≥30 - 100 |
75 |
75 |
75 |
150 |
150 |
150 |
150 |
150 |
300 |
300 |
|
>100 - 200 |
150 |
150 |
150 |
300 |
300 |
300 |
300 |
300 |
450 |
600 |
|
>200 - 300 |
150 |
150 |
225 |
300 |
300 |
450 |
450 |
450 |
600 |
|
|
>300 - 400 |
225 |
225 |
300 |
450 |
450 |
450 |
600 |
600 |
||
|
>400 - 500 |
225 |
300 |
450 |
450 |
600 |
600 |
||||
|
>500 - 600 |
300 |
300 |
450 |
600 |
600 |
|||||
|
>600 - 700 |
300 |
450 |
600 |
Administración cada 2 semanas: véase la tabla 4. |
||||||
|
Tabla 3. Administración cada 2 semanas. Dosis de XOLAIR® (mg por dosis) administrada por inyección subcutánea cada 2 semanas |
|||||||||||
|
IgE basal (UI/ml) |
Peso corporal (kg) |
||||||||||
|
>20-25 |
>25-30 |
>30–40 |
>40–50 |
>50–60 |
>60–70 |
>70–80 |
>80–90 |
>90– 125 |
>125–150 |
>150–200 |
|
|
≥30 - 100 |
Administración cada 4 semanas: Véase la tabla 3. |
225 |
|||||||||
|
>100 - 200 |
375 |
||||||||||
|
>200 - 300 |
375 |
525 |
|||||||||
|
>300 - 400 |
450 |
525 |
|||||||||
|
>400 - 500 |
375 |
375 |
525 |
600 |
|||||||
|
>500 - 600 |
375 |
450 |
450 |
600 |
|||||||
|
>600 - 700 |
225 |
375 |
450 |
450 |
525 |
||||||
|
>700 - 800 |
225 |
225 |
300 |
375 |
450 |
450 |
525 |
600 |
|||
|
>800 - 900 |
225 |
225 |
300 |
375 |
450 |
525 |
600 |
||||
|
>900 - 1000 |
225 |
300 |
375 |
450 |
525 |
600 |
NO ADMINISTRAR– no se dispone de datos para recomendar una dosis |
||||
|
>1000 - 1100 |
225 |
300 |
375 |
450 |
600 |
||||||
|
>1100 - 1200 |
300 |
300 |
450 |
525 |
600 |
||||||
|
>1200 - 1300 |
300 |
375 |
450 |
525 |
|||||||
|
>1300 - 1500 |
300 |
375 |
525 |
600 |
|||||||
Posología en la urticaria espontánea crónica (UEC): La dosis recomendada es de 300 mg, que se administra cada cuatro semanas por inyección subcutánea.
Se recomienda a los prescriptores que reevaluen periódicamente la necesidad de continuar el tratamiento.
La experiencia en ensayos clínicos del tratamiento a largo plazo durante más de 6 meses en esta indicación es limitada.
Poblaciones especiales:
• Disfunción renal o hepática: No se ha estudiado el efecto de la disfunción renal o hepática sobre la farmacocinética del omalizumab. Como la depuración del omalizumab, en dosis clínicas, depende principalmente del proceso de depuración de la IgG, por ejemplo, de su degradación en el sistema reticuloendotelial (SRE), es improbable que una disfunción renal o hepática pueda alterarla. Aunque no se recomienda ningún ajuste especial de la dosis, se debe tener cautela a la hora de administrar XOLAIR® a dichos pacientes (véase Advertencias y precauciones).
• Pacientes pediátricos: En el asma alérgica, no se ha determinado la inocuidad ni la eficacia del medicamento en pacientes menores de 6 años de edad; por consiguiente, no se recomienda el uso de XOLAIR® en tales pacientes.
En la urticaria espontánea crónica, no se ha determinado la inocuidad ni la eficacia del medicamento en pacientes menores de 12 años de edad.
• Pacientes geriátricos: Se dispone de escasa información sobre el uso de XOLAIR® en pacientes mayores de 65 años, pero no existen pruebas de que tales pacientes requieran una posología diferente de la de los pacientes adultos más jóvenes.
Forma de administración: Este producto debe administrarse únicamente por vía subcutánea. No debe administrarse por vía intravenosa ni intramuscular.
Las inyecciones se administran vía subcutánea en la región deltoidea del brazo. Si por alguna razón no pueden administrarse en esta zona, podrán administrarse alternativamente en el muslo.
Existe experiencia limitada con respecto a la autoadministración de XOLAIR®. Por lo tanto, está previsto que el tratamiento sea administrado únicamente por el profesional sanitario.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver Instrucciones de uso y manipulación.
INSTRUCCIONES DE USO Y MANIPULACIÓN:
XOLAIR®, vial de polvo y ampolla de disolvente para solución inyectable: El producto liofilizado necesita de 15 a 20 minutos para disolverse, pero en algunos casos puede requerir más tiempo. El producto completamente reconstituido es transparente o algo opalescente y puede presentar burbujas pequeñas o espuma en la superficie. Como el producto reconstituido es algo viscoso, hay que tener cuidado de extraer todo el producto del vial antes de eliminar el aire o el exceso de solución de la jeringa para obtener la dosis completa de 0,6 ml o 1,2 ml.
Para preparar XOLAIR® para la administración subcutánea, siga las siguientes indicaciones:
Viales de XOLAIR® 150 mg:
1. Extraiga 1,4 ml de agua para inyectables de la ampolla con una jeringa. La jeringa viene equipada de una aguja gruesa de calibre 18.
2. Con el vial boca arriba sobre una superficie plana, inserte la aguja y trasvase el agua para inyectables dentro del vial de omalizumab, aplicando técnicas asépticas convencionales, dirigiendo el agua para inyectables directamente sobre el polvo.
3. Con el vial siempre boca arriba, agítelo en círculos con vigor (sin sacudirlo) durante un minuto hasta que el polvo se humedezca de manera uniforme.
4. Para acelerar la disolución tras completar el paso 3, agite en círculos el vial colocado boca arriba, esta vez de forma más suave, por espacio de 5 o 10 segundos cada 5 minutos a fin de disolver el polvo restante.
* A veces pueden transcurrir más de 20 minutos antes de que el polvo se disuelva por completo. En tal caso, repita el paso 4 hasta que desaparezcan las partículas gelatinosas de la solución.
Una vez que el polvo se haya disuelto por completo, no deben quedar partículas gelatinosas en la solución. Las burbujas pequeñas o la espuma en la superficie del líquido son perfectamente normales. El producto reconstituido tiene un aspecto transparente o algo opalescente. No utilice el producto si observa partículas extrañas.
5. Coloque el vial boca abajo durante 15 segundos para que la solución fluya hacia el tapón.
Utilice una nueva jeringa de 3 centímetros cúbicos equipada de una aguja gruesa de calibre 18 e inserte la aguja en el vial invertido. Coloque el extremo de la aguja justo al final de la solución cuando extraiga la solución con la jeringa. Antes de extraer la aguja del vial, tire del émbolo y llévelo hasta el fondo del cilindro de la jeringa a efectos de extraer toda la solución del vial invertido.
6. Reemplace la aguja de calibre 18 por una de calibre 25 para inyección subcutánea.
7. Elimine el aire, las burbujas grandes y cualquier exceso de solución a fin de obtener la dosis requerida de 1,2 ml. Puede quedar una fina capa de pequeñas burbujas flotando en la superficie de la solución contenida en la jeringa. Como la solución es algo viscosa, la inyección puede durar entre 5 y 10 segundos. El vial proporciona 1,2 ml de XOLAIR® (150 mg de omalizumab).
8. Las inyecciones se administran por vía subcutánea en la región deltoide del brazo o en el muslo, evitando las lesiones urticariales.
El producto (XOLAIR® 150 mg, en polvo para solución inyectable) se suministra en viales para uso único y no contienen conservantes antibacterianos. Desde el punto de vista químico y físico, el producto reconstituido es estable durante 8 horas entre 2 y 8 °C y durante 4 horas a 30 °C. Desde el punto de vista microbiológico, el producto debe utilizarse de inmediato después de la reconstitución. Si no se usa de inmediato, tanto el tiempo de conservación durante el uso como las condiciones de conservación antes del uso son responsabilidad del usuario; por lo general, la solución no debe conservarse más de 8 horas entre 2 y 8 °C, a menos que se haya reconstituido en condiciones asépticas validadas y controladas. Todo producto no utilizado o material de desecho deberán eliminarse de conformidad con las normas locales.
Lugar de la inyección: El lugar de inyección es el sitio del cuerpo donde usted pondrá la inyección. XOLAIR® puede inyectarse en la parte superior externa del muslo o del brazo. Si es necesario efectuar varias inyecciones de una vez, repita la aplicación en el muslo o el brazo opuesto, evitando las lesiones urticariales.
Cómo usar la jeringa:
• Etapa 1: Sostenga la jeringa con la aguja apuntando hacia arriba y retire con cuidado el capuchón. No toque la aguja expuesta. A continuación, dé unos golpecitos suaves a la jeringa con el dedo para que suba la burbuja de aire. Empuje lentamente el émbolo para expulsar la burbuja de la jeringa, teniendo cuidado de no expulsar la solución accidentalmente.
• Etapa 2: Pellizque suavemente la piel del lugar de inyección. Inserte la aguja en el pliegue de la piel.
• Etapa 3: Sosteniendo la jeringa por el ala de sujeción, empuje lentamente el émbolo hacia abajo hasta haber inyectado toda la solución.
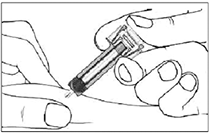
• Etapa 4: Una vez que se ha administrado la dosis completa, retire la aguja de la piel manteniendo el émbolo presionado.
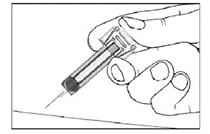
• Etapa 5: Suelte el émbolo lentamente y deje que el protector cubra automáticamente la aguja expuesta.
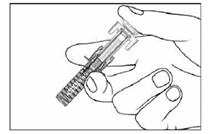
Nota: Si el protector de la aguja no se extiende automáticamente, presione firmemente el émbolo y luego suéltelo de manera que el protector cubra la aguja.
• Etapa 6: Deseche de inmediato la jeringa usada en un recipiente para objetos punzantes.
Vida útil: 48 meses.
Fabricante: Novartis Pharma Stein AG - Suiza
Revisión de texto: Información publicada en: Julio de 2013, corregida Diciembre de 2013
NOVARTIS PHARMA AG, Basilea (Suiza)
® Marca registrada
SOBREDOSIS: No se han notificado casos de sobredosis. No se ha determinado la dosis tolerada máxima de omalizumab (XOLAIR®). Se han administrado dosis únicas intravenosas de hasta 4000 mg a pacientes sin que se apreciaran signos de toxicidad limitante de la dosis. La mayor cantidad de fármaco a la que se expusieron los pacientes fue de 44 000 mg (dosis acumulada durante un período 20 semanas) y dicha cantidad no produjo ningún efecto adverso agudo.
En caso de sospecha de una sobredosis, se deberá monitorizar al paciente para cualquier signo o síntoma anormal. Se deberá buscar e instaurar tratamiento médico adecuado.
FARMACOLOGÍA CLÍNICA:
Farmacodinamia:
Características generales: Grupo farmacoterapéutico: fármacos para enfermedades obstructivas de las vías respiratorias, otros agentes sistémicos para enfermedades obstructivas de las vías respiratorias, código ATC: R03DX05.
El omalizumab es un anticuerpo monoclonal humanizado obtenido por ingeniería genética que se une selectivamente a la inmunoglobulina E (IgE) humana. El anticuerpo es una IgG1 (kappa) que contiene, enmarcadas por regiones humanas, las regiones determinantes de complementariedad de un anticuerpo murino que se fija a la IgE.
Pacientes con asma alérgica: La cascada alérgica se inicia cuando el alérgeno interconecta las IgE que se fijan a los receptores Fc?RI (receptores con gran afinidad por la IgE) en la superficie de los mastocitos y los basófilos. Ello produce la desgranulación de esas células efectoras y la liberación de histaminas, leucotrienos, citocinas y otros mediadores. Dichos mediadores guardan una relación causal con la fisiopatología del asma alérgica, que incluye la formación de un edema en las vías respiratorias, la contracción del músculo liso y una alteración de la actividad celular asociada al proceso inflamatorio. También contribuyen a producir los signos y síntomas de la enfermedad alérgica, a saber, broncoconstricción, producción de moco, sibilancias, disnea, opresión torácica, congestión nasal, estornudos, picazón nasal, rinorrea, picazón ocular y ojos llorosos.
El omalizumab se fija a la IgE e impide que ésta se una al receptor Fc?RI, reduciendo así la cantidad de IgE libre disponible para iniciar la cascada alérgica. El tratamiento con omalizumab de sujetos atópicos causó un pronunciado descenso del número de receptores Fc?RI. Además, la liberación in vitro de histamina de los basófilos aislados de individuos que habían recibido XOLAIR® disminuyó cerca del 90% tras la estimulación con un alérgeno, en comparación con los valores previos al tratamiento.
En los pacientes asmáticos de los estudios clínicos, las concentraciones séricas de IgE libre disminuyeron de forma dependiente de la dosis en la hora siguiente a la administración de la primera dosis y se mantuvieron constantes entre dos administraciones. La reducción media de las concentraciones séricas de IgE libre fue superior al 96% cuando se utilizaron las dosis recomendadas. Las concentraciones séricas de IgE total (es decir, unida y no unida a proteínas) aumentaron tras la primera dosis debido a la formación de complejos de omalizumab:IgE que se eliminan más lentamente que la IgE libre. Dieciséis semanas después de la primera dosis, las concentraciones séricas de IgE total eran, en promedio, unas cinco veces mayores que las cifras anteriores al tratamiento cuando se usaron ensayos convencionales. Tras interrumpir la administración de XOLAIR®, el aumento de la concentración de IgE total y la disminución de la concentración de IgE libre inducidos por XOLAIR® revirtieron sin que se observase ningún efecto de rebote en las concentraciones de IgE después del período de reposo farmacológico. Las concentraciones de IgE total no regresaron a sus niveles preterapéuticos sino hasta un año después de la retirada de XOLAIR®.
Pacientes con urticaria espontánea crónica (UEC): Existen varias teorías sobre la etiología de la UEC; una de ellas apunta a un origen autoinmunitario de la enfermedad. Se han aislado anticuerpos autoinmunitarios contra la IgE y su receptor, FceRI, del suero de algunos pacientes que padecían UEC. Tales autoanticuerpos pueden activar los basófilos o mastocitos y provocar la liberación de histamina.
Una de las hipótesis del modo de acción del omalizumab en la UEC es que reduce las concentraciones de IgE libre en la sangre y posteriormente en la piel. Ello causa un descenso del número de receptores superficiales de la IgE y de ese modo se reduce la consiguiente transcripción de señales a través de la vía del FceRI, lo cual redunda en una inhibición de las reacciones inflamatorias y de activación celular. Como corolario, disminuyen la frecuencia y la intensidad de los síntomas de UEC. Otra hipótesis es que la disminución de la cantidad circulante de IgE libre produce una desensibilización rápida e inespecífica de los mastocitos cutáneos. El descenso del número de FceRI puede contribuir a mantener la respuesta.
En los estudios clínicos de pacientes con UEC, el tratamiento con omalizumab produjo una reducción, dependiente de la dosis, de la cantidad de IgE libre y un aumento de la concentración de IgE total en el suero, de forma similar a lo que se observa en los pacientes con asma alérgica. La máxima disminución de IgE libre se observó 3 días después de la primera dosis subcutánea. Tras la administración repetida del medicamento cada 4 semanas, las cifras séricas de IgE libre anteriores a cada administración permanecieron estables entre las semanas 12 y 24 de tratamiento. Las cifras séricas de IgE total aumentaron después de la primera dosis debido a la formación de complejos omalizumab:IgE de eliminación más lenta que la IgE libre. Al cabo de 12 semanas de administración repetida del medicamento en dosis de entre 75 y 300 mg cada 4 semanas, las cifras séricas medias de IgE anteriores a la administración eran dos o tres veces mayores que las cifras determinadas antes de la terapia (preterapéuticas) y permanecieron estables entre las semanas 12 y 24 de tratamiento. Tras la retirada de XOLAIR®, durante un período de observación sin tratamiento de 16 semanas, las cifras de IgE libre aumentaron y las de IgE total disminuyeron hasta acercarse a los niveles preterapéuticos.
Farmacocinética:
Características generales:
• Absorción: Tras la administración subcutánea, el omalizumab se absorbe con una biodisponibilidad absoluta media del 62%. La farmacocinética de omalizumab es lineal a dosis mayores que 0,5 mg/kg.
Se obtienen curvas similares de concentración sérica (de omalizumab) y tiempo cuando se administran las formulaciones liofilizada o líquida de XOLAIR®.
• Distribución: In vitro, el omalizumab forma complejos de reducido tamaño con la IgE. Tanto in vitro como in vivo no se han observado complejos que precipitaran ni complejos con pesos moleculares superiores a 1x106 daltons.
Los estudios de distribución hística en macacos no evidenciaron una captación específica de I125-omalizumab por parte de ningún órgano o tejido.
• Eliminación: La depuración del omalizumab comprende procesos de depuración de IgG y de depuración a través de uniones específicas y de formación de complejos con su ligando específico, la IgE. La eliminación hepática de IgG incluye una degradación en las células endoteliales y reticuloendoteliales del hígado. La IgG inalterada se elimina asimismo en la bilis. En los estudios con ratones y monos, los complejos de omalizumab:IgE se eliminaron a través de interacciones con los receptores Fc? en el sistema reticuloendotelial del hígado a una velocidad generalmente mayor que la depuración de las IgG.
Pacientes con asma alérgica:
• Absorción: Después de administrar una dosis subcutánea única a pacientes adultos o adolescentes con asma, el omalizumab se absorbe con lentitud y alcanza su mayor concentración sérica al cabo de 7 u 8 días, en promedio. Tras la administración de dosis repetidas de omalizumab, el área bajo la curva de concentración sérica y tiempo desde el día 0 al 14 en el estado estacionario llegó a ser hasta seis veces mayor que la obtenida después de administrar la primera dosis.
• Distribución: Luego de la administración subcutánea, el volumen aparente de distribución de omalizumab en los pacientes asmáticos es de 78 ± 32 ml/kg.
• Eliminación: En los pacientes asmáticos, la semivida de eliminación sérica del omalizumab promedia los 26 días y su depuración aparente es de 2,4 ± 1,1 ml/kg/día, en promedio. La duplicación del peso corporal hace que la depuración aparente sea aproximadamente el doble.
• Edad, raza o grupo étnico, sexo biológico e índice de masa corporal: Se analizó la farmacocinética poblacional del omalizumab para determinar la influencia de las características demográficas. Los análisis de estos datos indican que no es necesario ajustar la dosis en los pacientes asmáticos en función de la edad (6–76 años), la raza, el grupo étnico, el sexo biológico o el índice de masa corporal.
Pacientes con urticaria espontánea crónica (UEC):
• Absorción: Tras la administración de una dosis subcutánea única a pacientes adultos o adolescentes con UEC, el omalizumab se absorbió con lentitud y alcanzó su mayor concentración sérica al cabo de 6 u 8 días, en promedio.
En pacientes con UEC, el omalizumab presenta una farmacocinética lineal en la gama de dosis de 75 a 600 mg administradas una sola vez por vía subcutánea. Luego de la administración de dosis de 75, 150 o 300 mg cada 4 semanas, la concentración sérica mínima de omalizumab aumenta de forma proporcional a la dosis.
• Distribución: Según un estudio de farmacocinética poblacional, la distribución de omalizumab en los pacientes con UEC es similar a la de los pacientes con asma alérgica.
• Eliminación: Las simulaciones farmacocinéticas poblacionales indicaron que, en los pacientes con UEC, la semivida de eliminación sérica del omalizumab promedia los 24 días en el estado estacionario y la depuración aparente en el estado estacionario, los 240 ml/día (que corresponde a 3,0 ml/kg/día en un paciente de 80 kg).
• Edad, raza o grupo étnico, sexo biológico, peso corporal, índice de masa corporal, IgE basal, autoanticuerpos anti-Fc?RI, comedicación: Los efectos de las covariables demográficas y de otros factores sobre la exposición al omalizumab se evaluaron mediante análisis de farmacocinética poblacional. También se evaluaron los efectos de covariables analizando la relación que existía entre las distintas concentraciones de omalizumab y las respuestas clínicas. Tales análisis han revelado que no hace falta ajustar la dosis en los pacientes con UEC en función de la edad (12 a 75 años), la raza o el grupo étnico, el sexo biológico, el peso corporal, el índice de masa corporal, la IgE basal, los autoanticuerpos anti-Fc?RI o el uso simultáneo de antihistamínicos H2 o de antagonistas de los receptores de leucotrienos (ARL).
• Pacientes con disfunción renal o hepática: No se dispone de datos farmacocinéticos ni farmacodinámicos en pacientes con disfunción renal o hepática aquejados de asma alérgica o UEC (véase Advertencias y precauciones).
CONSERVACIÓN: XOLAIR® debe conservarse en condiciones refrigeradas entre 2 y 8 °C. No congelar. Conservar el producto en el envase original.
El vial de polvo y la ampolla de disolvente de XOLAIR® pueden transportarse a temperatura ambiente controlada (?30°C) o entre 2 y 8 °C.
El período de validez ya incluye posibles desviaciones de la temperatura. Si fuera necesario, el producto puede volver a refrigerarse para uso posterior, pero ello no debe hacerse más de una vez.
Véase la caja plegable.
XOLAIR® no debe utilizarse después de la fecha de caducidad («EXP») indicada en el envase XOLAIR® debe conservarse fuera del alcance y de la vista de los niños.


