

YERVOY
IPILIMUMAB
Solución inyectable para infusión I.V.
1 Vial(es), 10 ml, 50 Miligramos
1 Vial monodosis, 40 ml, 200 Miligramos
FÓRMULA CUALICUANTITATIVA:
YERVOY® (ipilimumab) se suministra en frascos ampolla/viales de un solo uso de 50 mg/10 mL y 200 mg/40 mL. Cada frasco ampolla/vial de 10 mL contiene 50 mg de ipilimumab y excipientes c.s. Cada frasco ampolla/vial de 40 mL contiene 200 mg de ipilimumab y excipientes c.s. Cada mililitro contiene 5 mg de ipilimumab y los siguientes excipientes: tris clorhidrato, cloruro de sodio, manitol, ácido dietilentriaminopentaacético (DTPA), polisorbato 80 (de origen vegetal) y agua para inyección, USP; hidróxido de sodio y ácido clorhídrico, cantidad suficiente para pH 7.0.
PRESENTACIÓN/ALMACENAMIENTO Y MANIPULACIÓN:
YERVOY® se encuentra disponible en las siguientes presentaciones*:
• Un frasco ampolla/vial inyectable de 50 mg/10 mL (5 mg/mL), de un solo uso.
• Un frasco ampolla/vial inyectable de 200 mg/40 mL (5 mg/mL), de un solo uso.
Almacenar YERVOY® bajo refrigeración a una temperatura de 2 ºC a 8 ºC. No se debe congelar o agitar. Los frascos ampolla/viales deben protegerse de la luz. El tiempo de vida útil del producto es de 36 meses. No administrar el producto fuera de la fecha de vencimiento impreso en el rotulado.
*Alguna presentación puede no estar disponible en el mercado.
ACCIÓN TERAPÉUTICA:
Anticuerpo monoclonal humano recombinante que se une al antígeno 4 asociado a linfocitos T citotóxicos (CTLA-4). Inmunoglobulina IgGl K.
INDICACIONES Y USO:
Melanoma metastásico o no extirpable: YERVOY® está indicado para el tratamiento del melanoma metastásico o no extirpable [ver Estudios clínicos (Melanoma no extirpable o metastásico)].
Tratamiento adyuvante del melanoma: YERVOY® está indicado para el tratamiento adyuvante de pacientes con melanoma cutáneo con afectación patológica de los ganglios linfáticos regionales de más de 1 mm que han sido sometidos a resección completa, incluyendo linfadenectomía total [ver Estudios clínicos (Tratamiento adyuvante del melanoma)].
POSOLOGÍA/DOSIS Y ADMINISTRACIÓN:
Dosis recomendada para el melanoma metastásico o no extirpable: La dosis recomendada de YERVOY® es 3 mg/kg administrados por vía intravenosa durante 90 minutos, cada 3 semanas, por un máximo de 4 dosis. En el caso de toxicidad, las dosis se pueden retrasar pero todo el tratamiento debe de ser administrado dentro las 16 semanas de la primera dosis [ver Estudios clínicos (Melanoma no extirpable o metastásico)].
Dosis recomendada para el tratamiento adyuvante del melanoma: La dosis recomendada de YERVOY® es 10 mg/kg administrados por vía intravenosa durante 90 minutos, cada 3 semanas, por 4 dosis seguidos por 10 mg/kg cada 12 semanas por un máximo de 3 años [ver Estudios clínicos (Tratamiento adyuvante del melanoma)]. En el evento de toxicidad, las dosis se omiten, no se retrasan.
Modificaciones de la dosis recomendada:
Tabla 1: Modificaciones recomendadas del tratamiento para reacciones adversas de YERVOY® mediadas por respuesta inmunitaria
|
Sistema de órganos/ destino |
Reacciones adversas |
Modificación del tratamiento |
|
Endocrino |
Endocrinopatía sintomática |
Retener el uso de YERVOY®. Reanudar el uso de YERVOY® en pacientes con resolución de reacciones adversas parcial o completa (grado 0 a 1) y que están recibiendo menos de 7,5 mg de prednisona o equivalente por día. |
|
• Las reacciones sintomáticas demoran 6 semanas o más. • Incapacidad para reducir la dosis de corticosteroides a 7,5 mg de prednisona o equivalente por día. |
Descontinuar permanentemente el uso de YERVOY®. |
|
|
Oftalmológico |
Reacciones de grado 2 a 4 • No mejora a grado 1 dentro de 2 semanas mientras recibe terapia tópica o • Requiere tratamiento sistémico |
Descontinuar permanentemente el uso de YERVOY®. |
|
Todos los demás |
Grado 2 |
Retener el uso de YERVOY®. Reanudar el uso de YERVOY® en pacientes con resolución de reacciones adversas parcial o completa (grado 0 a 1) y que están recibiendo menos de 7,5 mg de prednisona o equivalente por día. |
|
• Reacciones de grado 2 que duran 6 semanas o más. • Incapacidad para reducir la dosis de corticosteroides a 7,5 mg de prednisona o equivalente por día. • Grado 3 o 4 |
Descontinuar permanentemente el uso de YERVOY®. |
Preparación y administración:
• No agitar el producto.
• Verificar visualmente que no haya partículas ni decoloración en los productos farmacológicos parenterales antes de la administración. Desechar el vial si la solución está turbia, presenta una decoloración pronunciada (el color de la solución puede ser amarillo pálido) o si presenta partículas extrañas que no sean transparentes o blancas, o partículas amorfas.
Preparación de la solución:.
• Dejar reposar los viales a temperatura ambiente durante, aproximadamente, 5 minutos antes de preparar la infusión.
• Extraer el volumen necesario de YERVOY® y colocarlo en una bolsa para infusión intravenosa.
• Diluir con cloruro de sodio para inyección al 0,9% o dextrosa al 5% para inyección, a fin de preparar una solución diluida con una concentración final que oscile entre 1 mg/mL a 2 mg/mL. Mezclar la solución diluida por inversión suave.
• Almacenar la solución diluida durante no más de 24 horas bajo refrigeración (2 ºC a 8 ºC) o a una temperatura entre 20 ºC a 25 ºC.
• Descartar la porción de solución no utilizada. Descartar los viales parcialmente usados o vacíos de YERVOY®. El hospital o centro de infusión debe seguir las normas establecidas para el manejo y eliminación de dichos viales. Instrucciones de administración.
• No mezclar YERVOY® con otros productos medicinales ni administrarlo como infusión junto con otros productos medicinales.
• Limpiar la vía intravenosa con cloruro de sodio para inyección al 0,9% o dextrosa para inyección al 5%, después de cada dosis.
• Administrar la solución diluida durante 90 minutos a través de una vía intravenosa que contenga un filtro en línea estéril, no pirogénico, con baja unión a proteínas (tamaño de poro de 0,2 µm a 1,2 µm).
• Administrar la solución diluida dentro de las 24 horas de su preparación, si ha sido almacenada bajo las condiciones señaladas en la sección previa [ver Preparación de la solución].
CONTRAINDICACIONES:
YERVOY® está contraindicado en pacientes con hipersensibilidad previamente demostrada a ipilimumab o a cualquier componente del producto.
REACCIONES ADVERSAS:
Las siguientes reacciones adversas se analizan en mayor detalle en otras secciones del prospecto.
• Enterocolitis mediada por la respuesta inmunitaria [ver Advertencias y precauciones (Enterocolitis mediada por la respuesta inmunitaria)].
• Hepatitis mediada por la respuesta inmunitaria [ver Advertencias y precauciones (Hepatitis mediada por la respuesta inmunitaria)].
• Dermatitis mediada por la respuesta inmunitaria [ver Advertencias y precauciones (Dermatitis mediada por la respuesta inmunitaria)].
• Neuropatías mediadas por la respuesta inmunitaria [ver Advertencias y precauciones (Neuropatías mediadas por la respuesta inmunitaria)].
• Endocrinopatías mediadas por la respuesta inmunitaria [ver Advertencias y precauciones (Endocrinopatías mediadas por la respuesta inmunitaria)].
• Otras reacciones adversas mediadas por la respuesta inmunitaria, incluidas las manifestaciones oculares [ver Advertencias y precauciones (Otras reacciones adversas mediadas por la respuesta inmunitaria, incluidas las manifestaciones oculares)]. En los pacientes que recibieron YERVOY® 3 mg/kg para el melanoma no extirpable o metastásico en el Estudio 1, 15% de los pacientes que recibieron monoterapia y el 12% de los pacientes tratados en combinación con la vacuna contra el péptido gp100 experimentaron reacciones mediadas por la respuesta inmunitaria de grado 3 a 5. En los pacientes que recibieron YERVOY® 10 mg/kg para el tratamiento adyuvante del melanoma en el Estudio 2, el 41% experimentó reacciones mediadas por la respuesta inmunitaria de grado 3 a 5.
Experiencia en estudios clínicos: Dado que los estudios clínicos se llevan a cabo en condiciones muy diferentes, los índices de reacciones adversas observados no pueden compararse en forma directa con los índices de otros estudios clínicos ni con la experiencia obtenida con tratamientos de la misma clase, y pueden no reflejar los índices observados en la práctica clínica. La información descrita a continuación refleja la exposición de 3 mg/kg de YERVOY® en el Estudio 1, un estudio aleatorizado en pacientes con melanoma no extirpable o metastásico y de 10 mg/kg de YERVOY® en el Estudio 2, un estudio aleatorizado en pacientes con resecado en etapa IIIA (>1 mm de implicación ganglionar), IIIB y IIIC (sin metástasis en tránsito) melanoma cutáneo. Reacciones adversas clínicamente significativas fueron evaluadas en un total de 982 pacientes tratados en los Estudios 1 y 2 y en 21 ensayos de rango de dosis (n=2478) que administraron YERVOY® en dosis de 0,1 a 20 mg/kg [ver Advertencias y precauciones (Otras reacciones adversas mediadas por la respuesta inmunitaria, incluidas las manifestaciones oculares)].
Melanoma no extirpable o metastásico: La seguridad de YERVOY® se evaluó en el Estudio 1, un estudio clínico aleatorizado, doble ciego, en el que 643 pacientes con melanoma no extirpable o metastásico previamente tratados recibieron 3 mg/kg de YERVOY® por 4 dosis administradas por infusión intravenosa como agente único (n=131), YERVOY® con una vacuna péptida gp100 bajo investigación (gp100) (n=380), o una vacuna péptida gp100 como agente único (n=132) [ver Estudios clínicos (Melanoma no extirpable o metastásico)]. Los pacientes en el estudio recibieron una mediana de 4 dosis (rango: 1 a 4 dosis). El estudio 1 excluyó a los pacientes con enfermedad autoinmune activa o a los que recibieron inmunosupresión sistémica para el trasplante de órganos. Las características de la población del estudio fueron: edad media de 57 años (rango: 19 a 90), 59% hombres, 94% blanco, y nivel de basal ECOG estado funcional 0 (56%). Se discontinuó la administración de YERVOY® a causa de las reacciones adversas en el 10% de los pacientes. La Tabla 2 incluye determinadas reacciones adversas observadas en el Estudio 1, que se presentaron en, al menos, el 5% de los pacientes en los grupos que recibían YERVOY® y que registraron un aumento de, al menos, el 5% en su incidencia en comparación con el grupo de control que recibía gp100, para los eventos de todos los grados, y un aumento en la incidencia de, al menos, el 1% con respecto al grupo de control para los eventos de grado 3 a 5.
Tabla 2: Determinadas reacciones adversas observadas en el Estudio 1
|
Porcentaje (%) de pacientesa |
||||||
|
YERVOY® 3 mg/kg |
YERVOY® 3 mg/ 100 kg + gp n=380 |
gp100 n=132 n=131 |
||||
|
Clasificación por sistema y órgano/término preferente |
Cualquier grado |
Grado 3 a 5 |
Cualquier grado |
Grado 3 a 5 |
Cualquier grado |
Grado 3 a 5 |
|
Trastornos generales y afecciones en el lugar de la administración |
||||||
|
Fatiga |
41 |
7 |
34 |
5 |
31 |
3 |
|
Trastornos gastrointestinales |
||||||
|
Diarrea |
32 |
5 |
37 |
4 |
20 |
1 |
|
Colitis |
8 |
5 |
5 |
3 |
2 |
0 |
|
Trastornos de la piel y el tejido subcutáneo |
||||||
|
Prurito |
31 |
0 |
21 |
<1 |
11 |
0 |
|
Erupción cutánea |
29 |
2 |
25 |
2 |
8 |
8 |
|
a Los índices de incidencia presentados en esta tabla se basan en los informes de eventos adversos, independientemente de la causalidad. |
||||||
La Tabla 3 incluye la incidencia por paciente de reacciones adversas graves, con riesgo de muerte o mortales mediadas por la respuesta inmunitaria obtenida a partir del Estudio I.
Tabla 3: Reacciones adversas graves o con riesgo de muerte mediadas por la respuesta inmunitaria, observadas en el Estudio 1
|
Porcentaje (%) de pacientes |
||
|
YERVOY® 3 mg/kg n=131 |
YERVOY® 3 mg/kg+gp100 n=380 |
|
|
Cualquier reacción adversa mediada por la respuesta inmunitaria |
15 |
12 |
|
Enterocolitisa,b |
7 |
7 |
|
Hepatotoxicidada |
1 |
2 |
|
Dermatitisa |
2 |
3 |
|
Neuropatíaa |
1 |
<1 |
|
Endocrinopatía |
4 |
1 |
|
Hipopituitarismo |
4 |
1 |
|
Insuficiencia suprarrenal |
0 |
1 |
|
Otras |
||
|
Neumonitis |
0 |
<1 |
|
Meningitis |
0 |
<1 |
|
Nefritis |
1 |
0 |
|
Eosinofiliac |
1 |
0 |
|
Pericarditisa,c |
0 |
<1 |
|
a Incluye desenlaces mortales. b Incluye la perforación intestinal. c No se establece la causa subyacente. |
||
Tratamiento adyuvante del melanoma: La seguridad de YERVOY® se evaluó en el Estudio 2, un estudio aleatorio (1:1), doble ciego, controlado con placebo en el que 945 pacientes con resecado en etapa IIIA (>1 mm de afectación ganglionar), IIIB y IIIC (sin metástasis en tránsito) melanoma cutáneo recibieron 10 mg/kg de YERVOY® (n=471) o placebo (n=474) administrado como una infusión intravenosa durante 4 dosis cada 3 semanas, seguidas de 10 mg/kg cada 12 semanas comenzando en la semana 24 hasta un máximo de 3 años [véase Estudios clínicos (Tratamiento adyuvante del melanoma)]. En este estudio, el 36% de los pacientes recibieron YERVOY® por más de 6 meses y el 26% de los pacientes recibieron YERVOY® por más de 1 año. Los pacientes tratados con YERVOY® en el estudio recibieron una mediana de 4 dosis (rango: 1 a 16). El Estudio 2 excluyó a los pacientes con tratamiento sistémico previo para el melanoma, enfermedad autoinmune, una enfermedad que requiera inmunosupresión sistémica, o un resultado positivo para la hepatitis B, hepatitis C, o VIH. Las características de la población del estudio fueron: edad media de 51 años (rango: 18 a 84 años), 62% varones, 99% blanco, y nivel de basal ECOG del estado de funcionamiento 0 (94%). YERVOY® se interrumpió por reacciones adversas en el 52% de los pacientes. La Tabla 4 presenta reacciones adversas seleccionadas del Estudio 2 que se produjeron en al menos el 5% de los pacientes tratados con YERVOY® y con al menos un 5% de mayor incidencia sobre el grupo placebo para eventos de todos los grados.
Tabla 4: Reacciones adversas seleccionadas del Estudio 2
|
Porcentaje (%) de pacientesa |
||||
|
YERVOY® 10 mg/kg n=471 |
Placebo n=474 |
|||
|
Clasificación por sistema y órgano/término preferente |
Cualquier grado |
Grado 3 a 5 |
Cualquier grado |
Grado 3 a 5 |
|
Trastornos de los tejidos cutáneos y subcutáneos |
||||
|
Erupción |
50 |
2,1 |
20 |
0 |
|
Prurito |
45 |
2,3 |
15 |
0 |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
49 |
10 |
30 |
2,1 |
|
Náusea |
25 |
0,2 |
18 |
0 |
|
Colitisb |
16 |
8 |
1,5 |
0,4 |
|
Vómitos |
13 |
0,4 |
6 |
0,2 |
|
Investigaciones |
||||
|
Pérdida de peso |
32 |
0.2 |
9 |
0.4 |
|
Trastornos generales y afecciones en el lugar de administración |
||||
|
Fatiga |
46 |
2,3 |
38 |
1,5 |
|
Pirexia |
18 |
1,1 |
4,9 |
0,2 |
|
Trastornos del sistema nervioso |
||||
|
Dolor de cabeza |
33 |
0,8 |
18 |
0,2 |
|
Trastornos del metabolismo y nutrición |
||||
|
Pérdida de apetito |
14 |
0,2 |
3,4 |
0,2 |
|
Trastornos psiquiátricos |
||||
|
Insomnio |
10 |
0 |
4,4 |
0 |
|
a Las incidencias presentadas en esta tabla están basadas en reportes de eventos adversos sin importar la causalidad. b Incluye 1 muerte. |
||||
La Tabla 5 presenta anormalidades del laboratorio seleccionadas del Estudio 2 que se presentaron en al menos el 10% de los pacientes tratados con YERVOY® en una mayor incidencia comparada con el placebo.
Tabla 5: Empeoramiento de las anormalidades del laboratorio que se presentaron en ≥10% de pacientes tratados con YERVOY® (Estudio 2)a
|
Test |
Porcentaje de pacientes con empeoramiento en las pruebas de laboratorio desde el nivel basala |
|||
|
YERVOY® |
Placebo |
|||
|
Todos los grados |
Grado 3 a 4 |
Todos los grados |
Grado 3 a 4 |
|
|
Química |
||||
|
Incremento de ALT |
46 |
10 |
16 |
0 |
|
Incremento de AST |
38 |
9 |
14 |
0,2 |
|
Incremento de lipasab |
26 |
9 |
17 |
4,5 |
|
Incremento de amilasab |
17 |
2,0 |
7 |
0,6 |
|
Incremento de la fosfatasa alcalina |
17 |
0,6 |
6 |
0,2 |
|
Incremento de bilirrubina |
11 |
1,5 |
9 |
0 |
|
Incremento de creatinina |
10 |
0,2 |
6 |
0 |
|
Hematología |
||||
|
Disminución de hemoglobina |
25 |
0,2 |
14 |
0 |
|
a Cada prueba de incidencia se basa en el número de pacientes que tenían ambas líneas de base y al menos mediciones de laboratorio disponibles durante el estudio. Excluyendo la lipasa y la amilasa, el grupo YERVOY® (rango: 466 a 470 pacientes) y el grupo placebo (rango: 472 a 474 pacientes). b Para la lipasa y la amilasa, el grupo YERVOY® (rango: 447 a 448 pacientes) y el grupo placebo (rango: 462 a 464 pacientes). |
||||
La Tabla 6 presenta la incidencia por paciente de las reacciones adversas mediadas por la respuesta inmunitaria graves, potencialmente mortales, o fatales del Estudio 2.
Tabla 6: Reacciones adversas mediadas por la respuesta inmunitaria severas a fatales del Estudio 2
|
Porcentaje (%) de Pacientes |
|
|
YERVOY® 10 mg/kg n=471 |
|
|
Cualquier reacción adversa mediada por la respuesta inmunitaria |
41 |
|
Enterocolitisa,b |
16 |
|
Hepatitis |
11 |
|
Dermatitis |
4,0 |
|
Neuropatíaa |
1,7 |
|
Endocrinopatía |
8 |
|
- Hipopituitarismo |
7 |
|
- Hipotiroidismo primario |
0,2 |
|
- Hipertiroidismo |
|
|
Otros |
|
|
Miocarditis |
0,2 |
|
Meningitis |
0,4 |
|
Pericarditis |
0,2 |
|
Neumonitis |
0,2 |
|
Uveítis |
0,2 |
|
a Incluyendo desenlace fatal. b Incluyendo perforación intestinal. c Etiología subyacente no establecida. |
|
Otras experiencias clínicas: En los estudios clínicos en los que se utilizaron dosis de YERVOY® de entre 0,3 a 10 mg/kg, también se informaron las siguientes reacciones adversas (incidencia menor del 1%, a menos que se especifique lo contrario): urticaria (2%), úlcera del intestino grueso, esofagitis, síndrome disneico agudo, insuficiencia renal y reacción a la infusión.
Experiencia posterior a la comercialización del producto: Las siguientes reacciones adversas se han identificado durante el uso de YERVOY® posterior a su aprobación. Dado que estas reacciones son reportadas voluntariamente por una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia ni establecer una relación causal con la exposición al fármaco.
Trastornos dérmicos y del tejido subcutáneo: Reacción medicamentosa con eosinofilia y síntomas sistémicos (síndrome DRESS).
Inmunogenicidad: Al igual que con todas las proteínas terapéuticas, existe un potencial de inmunogenicidad. Once (1,1%) de los 1.024 pacientes con melanoma no extirpable o metastásico evaluables tuvieron un resultado positivo en un ensayo de electroquimioluminiscencia (ECL) para anticuerpos de unión contra el ipilimumab emergentes al tratamiento (TE-ADAs). Este ensayo presentó limitaciones sustanciales para detectar los anticuerpos contra el ipilimumab en presencia del ipilimumab. Siete (4,9%) de 144 pacientes que recibieron ipilimumab y 7 (4,5%) de 156 pacientes que recibieron placebo para el tratamiento adyuvante del melanoma dieron un resultado positivo para TE-ADAs usando un ensayo de ECL con una mejor tolerancia a las drogas. Ninguno de los pacientes dieron positivo a anticuerpos neutralizantes. No se presentaron reacciones relacionadas a la infusión en pacientes que dieron positivo por TE-ADAs. Los resultados del ensayo de inmunogenicidad dependen en gran medida de varios factores, que incluyen: la sensibilidad y especificidad del ensayo, la metodología del ensayo, la manipulación de las muestras, el momento en que se extraen las muestras, los medicamentos concomitantes y las enfermedades subyacentes. Por estos motivos, la comparación de la incidencia de los anticuerpos contra ipilimumab con las incidencias de los anticuerpos contra otros productos puede ser engañosa.
INTERACCIONES MEDICAMENTOSAS:
No se han realizado estudios formales de interacción medicamentosa con YERVOY®.
ESTUDIOS CLÍNICOS:
Melanoma no extirpable o metastásico: La seguridad y la eficacia de YERVOY® se investigaron en un estudio aleatorizado (3:1:1), doble ciego, doble simulación (Estudio 1) que incluyó a 676 pacientes aleatorizados con melanoma no extirpable o metastásico tratados previamente con uno o más de los siguientes fármacos: aldesleukina, dacarbazina, temozolomida, fotemustina o carboplatino. De estos 676 pacientes, 403 fueron aleatorizados para recibir 3 mg/kg de YERVOY® en combinación con una vacuna péptida en investigación con adyuvantes incompletos de Freund (glicoproteína 100), 137 fueron aleatorizados para recibir 3 mg/kg de YERVOY®, y 136 fueron aleatorizados para recibir gp100 como agente único. El estudio enroló únicamente a pacientes con genotipo HLA-A2*0201; este genotipo HLA facilita la presentación inmune de la vacuna péptida en investigación. El estudio excluyó a los pacientes con enfermedad autoinmunitaria activa o a los pacientes que recibían inmunosupresión sistémica para el trasplante de órganos. Se administró YERVOY®/placebo en dosis de 3 mg/kg como infusión intravenosa cada 3 semanas con un total de 4 dosis. Se administró gp100/placebo en dosis de 2 mg de péptidos mediante inyecciones subcutáneas profundas cada 3 semanas con un total de 4 dosis. La evaluación de la respuesta tumoral se llevó a cabo en las semanas 12 y 24, y cada 3 meses a partir de la semana 24. Los pacientes con evidencia de respuesta tumoral objetiva a las 12 o 24 semanas fueron evaluados para confirmar la durabilidad de la respuesta a las 16 o 28 semanas, respectivamente. El criterio primario de valoración de la eficacia fue la sobrevida general (OS) en el grupo de tratamiento con YERVOY® más gp100 en comparación con el grupo de tratamiento con el único agente gp100. Los criterios secundarios de valoración de la eficacia fueron la OS en el grupo de tratamiento con YERVOY® más gp100 en comparación con el grupo de tratamiento con YERVOY®, la OS en el grupo de tratamiento con YERVOY® en comparación con el grupo de tratamiento con gp100, el índice de mejor respuesta global (BORR) en la semana 24 entre cada grupo de tratamiento del estudio y la duración de la respuesta. De los pacientes aleatorizados, el 61%, el 59%, y el 54% en los grupos de tratamiento con YERVOY® más gp100, YERVOY® y gp100, respectivamente, eran hombres. El 29% tenía ≥65 años, la mediana de la edad fue 57 años, el 71% se encontraba en el estadio M1c, el 12% tenía antecedentes de metástasis cerebral previamente tratada, el 98% tenía un estado general según el ECOG de 0 y 1, el 23% había recibido aldesleukina y el 38% presentaba un nivel elevado de LDH. El 61% de los pacientes aleatorizados a cualquiera de los grupos de tratamiento con YERVOY® recibió las 4 dosis programadas. La mediana de duración del seguimiento fue de 8,9 meses. Los resultados de la OS se muestran en la Tabla 7 y la Figura 1.
Tabla 7: Resultados de la sobrevida general
|
YERVOY® n=137 |
YERVOY® +gp100 n=403 |
gp100 n=136 |
|
|
Cociente de riesgo (vs. gp100) |
0,66 |
0,68 |
|
|
(IC del 95%) |
(0,51; 0,87) |
(0,55; 0,85) |
|
|
Valor de p |
p=0,0026a |
p=0,0004 |
|
|
Cociente de riesgo (vs. YERVOY®) |
1.04 |
||
|
(IC del 95%) |
(0,83; 1,30) |
||
|
Mediana (meses) |
10 |
10 |
6 |
|
(IC del 95%) |
(8,0; 13,8) |
(8,5; 11,5) |
(5,5; 8,7) |
|
a Valores no ajustados para múltiples comparaciones. |
|||
Figura 1: Sobrevida general
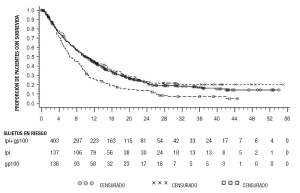
El índice de mejor respuesta global (BORR) evaluado por el investigador fue del 5,7% (IC del 95%: 3,7%; 8,4%) en el grupo de tratamiento con YERVOY® más gp100, del 10,9% (IC del 95%: 6,3%; 17,4%) en el grupo de tratamiento con YERVOY®, y del 1,5% (IC del 95%: 0,2%; 5,2%) en el grupo de tratamiento con gp100. La mediana de duración de la respuesta fue de 11,5 meses en el grupo de tratamiento con YERVOY® más gp100 y no se ha alcanzado en el grupo de tratamiento con YERVOY® o gp100.
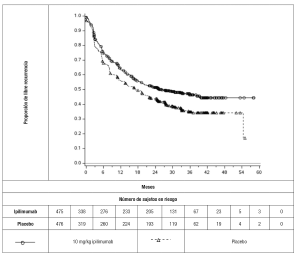
Tratamiento adyuvante del melanoma: La seguridad y eficacia de YERVOY® para el tratamiento adyuvante del melanoma fueron investigadas en el Estudio 2, un estudio aleatorizado (1:1), doble ciego, controlado con placebo en pacientes con resecado en la etapa IIIA (>1 mm afectación ganglionar), IIIB, y IIIC (sin metástasis en tránsito) melanoma cutáneo histológicamente confirmado. Los pacientes fueron aleatorizados para recibir YERVOY® 10 mg/kg o placebo en forma de infusión intravenosa cada 3 semanas por 4 dosis, seguidas de YERVOY® 10 mg/kg o placebo cada 12 semanas a partir de la semana 24 a la semana 156 (3 años) o hasta la recurrencia de la enfermedad documentada o toxicidad inaceptable. La inscripción requería la resección completa del melanoma con plena linfadenectomía dentro de 12 semanas antes de la aleatorización. Los pacientes con tratamiento previo para el melanoma, enfermedad autoinmune, y el uso previo o concomitante de agentes inmunosupresores eran inelegibles. La aleatorización se estratificó por etapas de acuerdo con El Comité Conjunto Estadounidense sobre el Cáncer (AJCC, en inglés) la clasificación de 2002 (Fase IIIA >1 mm de afectación ganglionar, etapa IIIB, etapa IIIC con 1 a 3 ganglios linfáticos involucrados y etapa IIIC con ≥4 ganglios linfáticos involucrados) y por regiones (América del Norte, Europa y Australia). Las principales medidas de resultados de eficacia fueron la supervivencia libre de recurrencia (RFS) definida como el tiempo entre la fecha de la aleatorización y la fecha de la primera recurrencia (metástasis local, regional o distante) o la muerte, lo que ocurra primero y evaluadas por un comité de revisión independiente y la supervivencia global. La evaluación del tumor se realizó cada 12 semanas durante los 3 primeros años, después, cada 24 semanas hasta la recurrencia a distancia. De los 951 pacientes incluidos, 475 fueron asignados al azar para recibir YERVOY® y 476 con placebo. La edad media fue de 51 años (rango: 18 a 84), el 62% eran hombres, el 99% eran blancos, el 94% tenían estado funcional ECOG de 0. En cuanto a la etapa de la enfermedad, el 20% tenía la Etapa IIIA con ganglios linfáticos >1 mm, el 44% tenía la Etapa IIIB, y el 36% tenía la Etapa IIIC (sin metástasis en tránsito). Otras características de la enfermedad de la población del estudio fueron: ganglios linfáticos clínicamente palpables (58%), 2 o más ganglios linfáticos positivos (54%) y las lesiones primarias ulceradas (42%). Los resultados RFS están en la Tabla 8 y la Figura 2. Basados en la observación de 282 muertes en el momento del análisis RFS, no se ha producido el análisis final de la supervivencia global (previsto en el momento de 491 muertes).
Tabla 8: Resultados de eficacia en el Estudio 2
|
Supervivencia libre de recurrencia |
YERVOY® N=475 |
Placebo N=476 |
|
Número de eventos, n (%) |
234 (49%) |
294 (62%) |
|
Recurrencia |
220 |
289 |
|
Muerte |
14 |
5 |
|
Promedio de meses |
26 |
17 |
|
(95% CI) |
(19, 39) |
(13, 22) |
|
Tasa de riesgo |
0.75 |
|
|
(95% CI) |
(0,64, 0,90) |
|
|
valor-p (rango logarítmico estratificadoa) |
p<0,002 |
|
|
a Estratificado por etapa de enfermedad |
||
Figura 2: Supervivencia libre de recurrencia
TOXICOLOGÍA NO CLÍNICA:
Carcinogénesis, mutagénesis, disfunción de la fertilidad: El potencial carcinogénico del ipilimumab no se ha evaluado en estudios en animales a largo plazo y el potencial genotóxico del ipilimumab no se ha evaluado. No se han llevado a cabo estudios de la fertilidad con ipilimumab.
ADVERTENCIA:
|
REACCIONES ADVERSAS MEDIADAS POR LA RESPUESTA INMUNITARIA YERVOY® puede provocar reacciones adversas graves y mortales mediadas por la respuesta inmunitaria. Estas reacciones mediadas por la respuesta inmunitaria pueden afectar cualquier sistema orgánico; sin embargo, las reacciones adversas graves más frecuentes mediadas por la respuesta inmunitaria son: enterocolitis, hepatitis, dermatitis (incluida la necrólisis epidérmica tóxica), neuropatía y endocrinopatía. La mayoría de estas reacciones mediadas por la respuesta inmunitaria se manifestaron inicialmente durante el tratamiento; sin embargo, la menor parte de estos eventos se presentó algunas semanas o algunos meses después de haber interrumpido la administración de YERVOY®. Discontinuar la administración de YERVOY® en forma permanente e iniciar un tratamiento con dosis altas de corticosteroides sistémicos para tratar las reacciones graves mediadas por la respuesta inmunitaria [ver Posología/Dosis y Administración (Modificaciones de la dosis recomendada.)]. Evaluar a los pacientes para detectar signos y síntomas de enterocolitis, dermatitis, neuropatía y endocrinopatía, y evaluar los análisis de química clínica, incluyendo pruebas de función hepática, nivel de hormona adrenocorticotrópica (ACTH) y pruebas de función tiroidea en el nivel basal y antes de cada dosis [ver Advertencias y Precauciones (Enterocolitis mediada por la respuesta inmunitaria, Hepatitis mediada por la respuesta inmunitaria, Dermatitis mediada por la respuesta inmunitaria, Neuropatías mediadas por la respuesta inmunitaria, Endocrinopatías mediadas por la respuesta inmunitaria.)]. |
Nota: Se han omitido secciones, subsecciones y/o tablas numeradas de la información completa sobre prescripción por no ser aplicables.
ADVERTENCIAS Y PRECAUCIONES:
YERVOY® puede provocar reacciones graves y mortales mediadas por la respuesta inmunitaria [ver Recuadro de advertencias].
Enterocolitis mediada por la respuesta inmunitaria: Puede ocurrir enterocolitis mediada por la respuesta inmunitaria, incluyendo casos fatales, al usar YERVOY®. Se debe controlar a los pacientes para detectar signos y síntomas de enterocolitis (como diarrea, dolor abdominal, moco o sangre en las heces, con o sin fiebre) y de perforación intestinal (como íleo y signos peritoneales). En los pacientes sintomáticos, se deben descartar las causas infecciosas y se debe considerar una evaluación endoscópica en el caso de los síntomas persistentes o graves. Discontinuar la administración de YERVOY® en forma permanente en los pacientes que presentan enterocolitis grave y comenzar el tratamiento con corticosteroides sistémicos a una dosis de 1 a 2 mg/kg/día de prednisona o una sustancia equivalente. Una vez que el paciente mejore y presente el grado 1 o un grado menor, se debe comenzar a disminuir progresivamente la administración de corticosteroides y continuar disminuyendo esta administración durante, al menos, 1 mes. En estudios clínicos, la disminución progresiva de la administración de corticosteroides provocó, en algunos pacientes, la recurrencia o el empeoramiento de los síntomas de la enterocolitis. Considerar añadir anti-TNF u otros agentes inmunosupresores para el tratamiento de la enterocolitis mediada por la respuesta inmunitaria que no responda a corticosteroides sistémicos dentro de 3 a 5 días o que sea recurrente después de la mejoría de síntomas. Se debe interrumpir el tratamiento con YERVOY® si el paciente presenta enterocolitis moderada; se debe administrar un tratamiento antidiarreico y, si la afección continúa durante más de 1 semana, se debe iniciar un tratamiento con corticosteroides sistémicos a una dosis de 0,5 mg/kg/día de prednisona o una sustancia equivalente [ver Posología/Dosis y Administración (Modificaciones de la dosis recomendada)].
Melanoma metastásico: En pacientes que fueron tratados con YERVOY® 3 mg/kg en el Estudio 1, hubo 34 casos (7%) de enterocolitis mediada por la respuesta inmunitaria en que fueron graves, con riesgo de muerte o mortales (diarrea con 7 o más deposiciones sobre el nivel basal, fiebre, íleo y signos peritoneales; grados 3 a 5). Asimismo, 28 pacientes (5%) que recibían tratamiento con YERVOY® presentaron enterocolitis moderada (diarrea con hasta 6 deposiciones sobre el nivel basal, dolor abdominal, moco o sangre en las deposiciones; grado 2). Entre todos los pacientes que recibieron tratamiento con YERVOY® (n=511), 5 pacientes (1%) desarrollaron perforación intestinal, 4 pacientes (0,8%) murieron a causa de complicaciones, y 26 pacientes (5%) fueron hospitalizados a causa de una enterocolitis grave. La mediana del tiempo hasta el inicio de la enterocolitis de grado 3 a 5 fue de 1,7 meses (rango: 11 días a 3,1 meses) y en pacientes con enterocolitis de grado 2 fue de 1,4 meses (rango: 2 días a 4,3 meses). Veintinueve pacientes (85%) con enterocolitis de grado 3 a 5 recibieron tratamiento con dosis altas de corticosteroides (≥40 mg de prednisona o un equivalente al día), con una dosis media de 80 mg/día de prednisona o un equivalente; la mediana de duración del tratamiento fue de 16 días (se extendió hasta 3,2 meses) y luego la administración de corticosteroides se redujo progresivamente. Entre los 28 pacientes con enterocolitis moderada, el 46% no recibió corticosteroides sistémicos, el 29% recibió tratamiento con <40 mg de prednisona o una sustancia equivalente al día durante una mediana de duración de 1,2 meses, y el 25% de los pacientes fueron tratados con dosis altas de corticosteroides durante una mediana de tiempo de 10 días antes de reducir progresivamente la administración de corticosteroides. Se administró infliximab a 5 (8%) de los 62 pacientes que presentaron enterocolitis moderada, grave o con riesgo de muerte, mediada por la respuesta inmunitaria luego de una respuesta inadecuada a los corticosteroides. De los 34 pacientes con enterocolitis de grado 3 a 5, el 74% presentó una resolución completa, el 3% presentó una mejoría a grado 2, y el 24% no presentó mejoría. Entre los 28 pacientes con enterocolitis de grado 2, el 79% presentó una resolución completa, el 11% presentó una mejoría, y el 11% no presentó mejoría.
Tratamiento adyuvante del melanoma: En pacientes que fueron tratados con 10 mg/kg de YERVOY® en el Estudio 2, la enterocolitis mediada por la respuesta inmunitaria de grado 3 a 5 se presentó en 76 pacientes (16%) y la de grado 2 se presentó en 68 pacientes (14%). Siete pacientes (1,5%) desarrollaron perforaciones intestinales y 3 pacientes (0,6%) murieron a causa de complicaciones [ver Reacciones adversas (Experiencia en estudios clínicos)].
La mediana del tiempo hasta el inicio de la enterocolitis de grado 3 a 4 fue de 1,1 meses (rango: 1 día a 33,1 meses) y para la enterocolitis de grado 2 fue de 1,1 meses (rango: 1 día a 20,6 meses). Setenta y un pacientes (95%) con enterocolitis de grado 3 a 4 fueron tratados con corticosteroides sistémicos. La mediana de la duración del tratamiento fue de 4,7 meses (rango de hasta 52,3 meses). De los 68 pacientes con enterecolitis moderada, 51 pacientes (75%) fueron tratados con corticosteroides sistémicos con una mediana de duración del tratamiento de 3,5 meses (rango de hasta 52,2 meses). Inmunosupresores no corticoides, que consisten casi exclusivamente de infliximab, fueron usados para tratar el 36% de los pacientes con enterocolitis de grado 3 a 4 y el 15% de los pacientes con enterocolitis de grado 2. De los 75 pacientes con enterocolitis mediada por la respuesta inmunitaria de grado 3 a 4, el 86 % experimentó una resolución completa, el 3% experimentó una mejoría al grado 1, y el 11% no mejoró. De los 68 pacientes con enterocolitis de grado 2, el 94% experimentó una resolución completa, el 3 % experimentó una mejoría al grado 1, y el 3% no mejoró.
Hepatitis mediada por la respuesta inmunitaria: La hepatitis mediada por la respuesta inmunitaria, incluyendo casos fatales, puede ocurrir con el uso de YERVOY®. Supervisar las pruebas de función hepática (niveles de transaminasas hepáticas y bilirrubina) y evaluar a los pacientes para detectar signos y síntomas de la hepatotoxicidad antes de cada dosis de YERVOY®. En los pacientes con hepatotoxicidad, descartar causas infecciosas o malignas y aumentar la frecuencia del monitoreo de pruebas de la función hepática hasta la resolución. Se debe descontinuar la administración de YERVOY® de modo permanente en los pacientes con hepatotoxicidad de grado 3 a 4, y se debe administrar un tratamiento con corticosteroides sistémicos a una dosis de 1 a 2 mg/kg/día de prednisona o una sustancia equivalente. Cuando las pruebas de función hepática muestren una mejoría sostenida o los valores regresen al nivel basal, se debe comenzar a reducir en forma progresiva la administración de corticosteroides durante 1 mes. En el programa de desarrollo clínico de YERVOY®, se administró el tratamiento con micofenolato en los pacientes que presentaban hepatitis grave persistente a pesar de las dosis altas de corticosteroides. Se debe interrumpir la administración de YERVOY® en los pacientes que presentan una hepatotoxicidad de grado 2 [ver Posología/Dosis y administración (Modificaciones de la dosis recomendada)].
Melanoma metastásico: En el Estudio 1, 8 pacientes (2%) tratados con YERVOY® 3 mg/kg presentaron una hepatotoxicidad grave, con riesgo de muerte o mortal (elevaciones de la AST o la ALT o más de 5 veces superiores al límite superior normal o elevaciones en los niveles de bilirrubina total más de 3 veces superiores al límite superior normal; grado 3 a 5). El 0,2% de los pacientes que recibieron tratamiento con YERVOY® presentaron una insuficiencia hepática mortal, y el 0,4% de los pacientes tuvieron que ser hospitalizados. Otros 13 pacientes (2,5%) presentaron una hepatotoxicidad moderada que se manifestó en anomalías en las pruebas de la función hepática (elevaciones de la AST o la ALT de más de 2,5 veces, aunque no más de 5, del límite superior normal, o una elevación de la bilirrubina total de más de 1,5 veces, aunque no más de 3, del límite superior normal; grado 2). No se confirmó patología subyacente en todos los pacientes, sin embargo en algunos casos la biopsia confirmó la hepatitis mediada por la respuesta inmunitaria. No hubo una cantidad suficiente de pacientes cuya hepatitis haya sido confirmada mediante biopsia, a fin de caracterizar la evolución clínica de este evento.
Tratamiento adyuvante del melanoma: En los pacientes que recibieron YERVOY® 10 mg/kg en el Estudio 2, la hepatitis mediada por la respuesta inmunitaria de grado 3 a 4 se presentó en 51 pacientes (11%) y la hepatitis moderada mediada por la respuesta inmunitaria de grado 2 se presentó en 22 pacientes (5%). La biopsia de hígado realizada en 6 pacientes con grado 3 a 4 de hepatitis mostró evidencia de hepatitis tóxica o autoinmune. La mediana de tiempo hasta la aparición de grado 3 a 4 de la hepatitis fue de 2,0 meses (rango: 1 día a 4,2 meses) y para el grado 2 de la hepatitis fue de 1,4 meses (rango: 13 días a 6,5 meses). De los 51 pacientes con grado 3 a 4 de la hepatitis mediada por la respuesta inmunitaria, el 94% experimentó una resolución completa, el 4% experimentó una mejoría al grado 1, y el 2% no mejoró. De los 22 pacientes con grado 2 de la hepatitis mediada por la respuesta inmunitaria, el 91% experimentó una resolución completa y el 9% no mejoró. Cuarenta y seis pacientes (90%) con grado 3 a 4 de la hepatitis fueron tratados con corticosteroides sistémicos. La mediana de duración del tratamiento fue de 4,4 meses (rango de hasta 56,1 meses). Dieciséis pacientes (73%) con hepatitis moderada fueron tratados con corticosteroides sistémicos. La mediana de duración del tratamiento fue de 2,6 meses (rango de hasta 41,4 meses).
Administración concurrente con vemurafenib: En un ensayo de hallazgo de dosis, se produjeron aumentos de Grado 3 en las transaminasas, con o sin aumentos concomitantes de la bilirrubina total, en 6 de 10 pacientes que recibieron YERVOY® (3 mg/kg) y vemurafenib (960 mg dos veces por día o 720 mg dos veces por día) en forma concurrente.
Dermatitis mediada por la respuesta inmunitaria: La dermatitis mediada por la respuesta inmunitaria, incluyendo casos fatales, puede producirse con el uso de YERVOY®. Controlar a los pacientes para detectar signos y síntomas de la dermatitis, como la erupción cutánea y el prurito. A menos que una etiología alternativa haya sido identificada, los signos o síntomas de la dermatitis deben considerarse mediados por la respuesta inmunitaria. Descontinuar de manera permanente el uso de YERVOY® en pacientes con el síndrome de Stevens-Johnson, necrólisis epidérmica tóxica o erupción cutánea complicada por úlceras dérmicas de espesor total, o manifestaciones necróticas, ampollosas o hemorrágicas. Administrar los corticosteroides sistémicos a una dosis de 1 a 2 mg/kg/día de prednisona o sustancia equivalente. Cuando se logre controlar la dermatitis, la reducción progresiva de corticosteroides debe darse durante un período de al menos 1 mes. Retener el uso de la dosis de YERVOY® en pacientes con signos y síntomas de moderados a graves [ver Posología/ Dosis y administración (Modificaciones de la dosis recomendada)]. Para la dermatitis leve a moderada, tal como la erupción cutánea localizada y el prurito, tratar sintomáticamente. Administrar corticosteroides tópicos o sistémicos si no hay mejoría de los síntomas dentro de 1 semana.
Melanoma metastásico. De los pacientes que recibieron YERVOY® 3 mg/kg en el Estudio 1, 13 pacientes (2,5%) tratados con YERVOY® presentaron dermatitis grave, con riesgo de muerte o mortal mediada por la respuesta inmunitaria (p. ej., el síndrome de Stevens-Johnson, necrólisis epidérmica tóxica o erupción cutánea complicada por úlceras dérmicas de espesor total, o manifestaciones necróticas, ampollosas o hemorrágicas; grados 3 a 5). Un paciente (0,2%) murió como consecuencia de una necrólisis epidérmica tóxica, y otro paciente debió ser hospitalizado a causa de una dermatitis grave. Sesenta y tres pacientes (12%) presentaron dermatitis moderada (grado 2). La mediana del tiempo hasta el inicio de la dermatitis moderada, grave o con riesgo de muerte, mediada por la respuesta inmunitaria, fue de 22 días y se extendió hasta 4,0 meses a partir del inicio de la administración de YERVOY®. Siete pacientes (54%) tratados con YERVOY® que presentaron dermatitis grave recibieron dosis altas de corticosteroides (mediana de la dosis de 60 mg de prednisona/día o una sustancia equivalente) durante 3,4 meses, como máximo, seguido por la disminución gradual de la administración de corticosteroides. De estos 7 pacientes, 6 tuvieron una resolución completa; el tiempo hasta la mejoría completa fue de hasta 3,6 meses. Entre estos 63 pacientes que presentaban dermatitis moderada, 25 (40%) recibieron tratamiento con corticosteroides sistémicos (mediana de 60 mg/día de prednisona o una sustancia equivalente) durante una mediana de 15 días; 7 pacientes (11%) recibieron tratamiento únicamente con corticosteroides tópicos, y 31 pacientes (49%) no recibieron ni corticosteroides sistémicos ni tópicos. Se informó que 44 pacientes (70%) que presentaron dermatitis moderada tuvieron una mejoría completa, 7 (11%) mejoraron y presentaron una intensidad leve (grado 1), y 12 (19%) no informaron mejorías.
Tratamiento adyuvante del melanoma: En pacientes que recibían 10 mg/kg de YERVOY® en el Estudio 2, la dermatitis mediada por la respuesta inmune de grado 3 a 4 se presentó en 19 pacientes (4%). Hubo 99 pacientes (21%) con dermatitis moderada (grado 2). La mediana del tiempo hasta el inicio de la dermatitis de grado 3 a 4 fue de 14 días (rango: 5 días a 11,3 meses) y la de dermatitis de grado 2 fue de 11 días (rango: 1 día a 16,6 meses). Fueron tratados 16 pacientes (84%) con dermatitis de grado 3 a 4 con corticosteroides sistémicos por un promedio de 21 días (rango de hasta 49,2 meses) resultando en una completa resolución de dermatitis en una mediana de tiempo de 4,3 meses (rango de hasta 44,4 meses). De los 3 pacientes (16%) que no fueron tratados con corticosteroides sistémicos o tópicos, 2 de ellos (11%) tuvieron una completa resolución y 1 tuvo una mejoría al grado 1. De los 99 pacientes con dermatitis de grado 2, 67 (68%) fueron tratados con corticosteroides sistémicos por un promedio de 2,6 meses, 16 (16%) fueron tratados solo con corticosteroides tópicos, y 16 (16%) no recibieron corticosteroides sistémicos o tópicos. Setenta y siete pacientes (78%) tuvieron una completa resolución, 15 (15%) mejoraron a severidad leve (grado 1), y 7 (7%) no presentaron mejoría.
Neuropatías mediadas por la respuesta inmunitaria: Las neuropatías mediadas por la respuesta inmunitaria, incluyendo casos fatales, pueden ser producidas al usar YERVOY®. Controlar a los pacientes para detectar síntomas de neuropatía motora o sensorial, como debilidad unilateral o bilateral, alteraciones sensoriales o parestesia. Se debe discontinuar la administración de YERVOY® en forma permanente en los pacientes que presentan neuropatía grave (que interfiere en sus actividades diarias), como síndromes parecidos al de Guillain-Barré. Se debe administrar un tratamiento, según corresponda, para la neuropatía grave. Debe considerarse comenzar a administrar corticosteroides sistémicos a una dosis de 1 a 2 mg/kg/día de prednisona o una sustancia equivalente para el tratamiento de la neuropatía grave. Se debe interrumpir la dosis de YERVOY® en los pacientes que presentan neuropatía moderada (que no interfiere en sus actividades diarias) [ver Posología/Dosis y Administración (Modificaciones de la dosis recomendada)].
Melanoma metastásico: En los pacientes que reciben 3 mg/kg de YERVOY® en el Estudio 1, se reportaron 1 caso fatal de síndrome de Guillain-Barré y 1 caso de neuropatía motora periférica severa (grado 3). A través del programa de desarrollo clínico de YERVOY® se ha reportado miastenia gravis y casos adicionales del síndrome de Guillain-Barré.
Tratamiento adyuvante del melanoma: En los pacientes que recibieron 10 mg/kg de YERVOY® en el Estudio 2, la neuropatía mediada por la respuesta inmunitaria de grado 3 a 5 se presentó en 8 pacientes (2%); la única víctima mortal se debió a complicaciones del síndrome de Guillain-Barré [ver Reacciones adversas (Experiencia en estudios clínicos)]. La neuropatía mediada por la respuesta inmunitaria moderada de grado 2 se presentó en 1 paciente (0,2%). El tiempo de aparición entre los 9 pacientes con grado 2 a 5 de neuropatía mediada por la respuesta inmunitaria varió de 1,4 a 27,4 meses. Los 8 pacientes con grado 3 a 5 de neuropatía fueron tratados con corticosteroides sistémicos (rango: 3 días a 38,3 meses) y 3 recibieron también tacrolimus. Cuatro de los 8 pacientes con grado 3 a 5 de neuropatía mediada por la respuesta inmunitaria experimentaron resolución completa, 1 mejoró a grado 1, y 3 no presentaron mejoras. El único paciente con grado 2 de neuropatía mediada por la respuesta inmunitaria experimentó resolución completa sin el uso de corticosteroides.
Endocrinopatías mediadas por la respuesta inmunitaria: Endocrinopatías mediadas por la respuesta inmunitaria, incluyendo casos mortales, pueden darse con el uso de YERVOY®. Controlar a los pacientes para detectar signos y síntomas clínicos de hipofisitis, insuficiencia suprarrenal (incluyendo crisis suprarrenal), y la hiper- o hipotiroidismo. Los pacientes pueden presentar fatiga, dolor de cabeza, cambios en el estado mental, dolor abdominal, hábitos intestinales inusuales e hipotensión, o síntomas inespecíficos que pueden parecerse a los de otras causas como metástasis cerebral o enfermedad subyacente. A menos que una etiología alternativa haya sido identificada, los signos o síntomas de endocrinopatías deben considerarse mediados por la respuesta inmunitaria. Monitorear pruebas de química clínica, el nivel de la hormona adrenocorticotrópica (ACTH), y pruebas de función tiroidea al inicio del tratamiento, antes de cada dosis, y como sea indicado clínicamente basado en los síntomas. En un número limitado de pacientes, la hipofisitis fue diagnosticada por estudios de imagen a través de la ampliación de la glándula pituitaria. Retener la dosificación de YERVOY® en pacientes sintomáticos y considerar la derivación a un endocrinólogo. Iniciar corticosteroides sistémicos en una dosis de 1 a 2 mg/kg/día de prednisona o sustancia equivalente, e iniciar una terapia de reemplazo hormonal apropiado [ver Posología/Dosis y Administración (Modificaciones de la dosis recomendada)].
Melanoma metastásico: De los pacientes que recibieron YERVOY® 3 mg/kg en el Estudio 1, 9 pacientes (1,8%) tratados con YERVOY® presentaron endocrinopatías graves o con riesgo de muerte mediadas por la respuesta inmunitaria (que requirieron hospitalización, tratamiento médico urgente o que interfirieron en sus actividades diarias; grado 3 a 4). Estos 9 pacientes presentaron hipopituitarismo, y algunos tuvieron algunas endocrinopatías concomitantes adicionales, como insuficiencia suprarrenal, hipogonadismo e hipotiroidismo. Seis de los nueve pacientes fueron hospitalizados a causa de endocrinopatías graves. Doce pacientes (2,3%) presentaron endocrinopatías moderadas (que requirieron reemplazo hormonal o tratamiento médico; grado 2). Estas endocrinopatías incluyeron: hipotiroidismo, insuficiencia suprarrenal, hipopituitarismo y 1 caso de hipertiroidismo y 1 caso de síndrome de Cushing. La mediana del tiempo hasta el inicio de la endocrinopatía de moderada a grave mediada por la respuesta inmunitaria fue de 2,5 meses y se extendió hasta 4,4 meses después de haber comenzado el tratamiento con YERVOY®. De los 21 pacientes con endocrinopatía moderada o con riesgo de muerte, 17 pacientes necesitaron un tratamiento de reemplazo hormonal a largo plazo que incluyó, en la mayoría de los casos, hormonas suprarrenales (n=10) y hormonas tiroideas (n=13).
Tratamiento adyuvante del melanoma: En los pacientes que recibieron YERVOY® 10 mg/kg en el Estudio 2, las endocrinopatías mediadas por la respuesta inmunitaria de grado 3 a 4 se presentaron en 39 pacientes (8%), y de grado 2 en 93 pacientes (20%). De los 39 pacientes con grado 3 a 4 de endocrinopatías mediadas por la respuesta inmunitaria, 35 pacientes tenían hipopituitarismo (asociado con una o más endocrinopatías secundarias, por ejemplo, insuficiencia suprarrenal, hipogonadismo, e hipotiroidismo), 3 pacientes tenían hipertiroidismo, y 1 paciente tenía hipotiroidismo primario. La mediana de tiempo hasta la aparición de la endocrinopatía de grado 3 a 4 fue de 2,2 meses (rango: 2 días a 8 meses). Veintisiete de los 39 pacientes (69%) fueron hospitalizados por endocrinopatías mediadas por la respuesta inmunitaria, y se reportaron 4 pacientes (10%) que tuvieron resolución. De los 93 pacientes con grado 2 de endocrinopatía mediada por la respuesta inmunitaria, 74 tenían hipopituitarismo primario (asociado con uno o más endocrinopatía secundaria, por ejemplo, insuficiencia suprarrenal, hipogonadismo, e hipotiroidismo), 9 tenían hipotiroidismo primario, 3 tenían hipertiroidismo, 3 tenían tiroiditis con hipo- o hipertiroidismo, 2 tenían hipogonadismo, 1 tenía tanto el hipertiroidismo como el hipopituitarismo, y 1 paciente desarrolló oftalmopatía de Graves. La mediana de tiempo hasta la aparición de grado 2 de endocrinopatía mediada por la respuesta inmunitaria fue de 2,1 meses (rango: 9 días a 19,3 meses), y un 20% fue reportado con resolución. Ciento veinticuatro pacientes recibieron corticosteroides sistémicos como terapia de inmunosupresión y/o reemplazo de la hormona suprarrenal para la endocrinopatía mediada por la respuesta inmunitaria de grado 2 a 4. De ellos, 42 (34%) fueron capaces de interrumpir los corticosteroides. Setenta y tres pacientes recibieron hormonas tiroideas para el tratamiento de hipotiroidismo mediado por la respuesta inmunitaria de grado 2 a 4. De ellos, 14 pacientes (19%) fueron capaces de interrumpir la terapia de reemplazo de tiroides.
Otras reacciones adversas mediadas por la respuesta inmunitaria, incluidas las manifestaciones oculares: Se debe discontinuar la administración de YERVOY® en forma permanente si se presentan reacciones adversas clínicamente significativas o reacciones adversas graves mediadas por la respuesta inmunitaria. Se debe iniciar la administración de corticosteroides sistémicos a una dosis de 1 a 2 mg/kg/día de prednisona o una sustancia equivalente para el tratamiento de las reacciones adversas graves mediadas por la respuesta inmunitaria. Se deben administrar gotas oftálmicas con corticosteroides a los pacientes que desarrollan uveítis, iritis o epiescleritis. Se debe discontinuar la administración de YERVOY® en forma permanente para el tratamiento de las enfermedades oculares mediadas por la respuesta inmunitaria que no responden al tratamiento inmunosupresor local [ver Posología/Dosis y administración (Modificaciones de la dosis recomendada)].
Melanoma metastásico: En el Estudio 1, las siguientes reacciones adversas mediadas por la respuesta inmunitaria clínicamente significativas se observaron en menos del 1% de los pacientes tratados con YERVOY®: nefritis, neumonitis, meningitis, pericarditis, uveítis, iritis y anemia hemolítica.
Tratamiento adyuvante del melanoma: En el Estudio 2, las siguientes reacciones adversas mediadas por la respuesta inmunitaria clínicamente significativas se observaron en menos del 1% de los pacientes tratados con YERVOY® a menos que se especifique: eosinofilia (2,1%), pancreatitis (1,3%), meningitis, neumonía, sarcoidosis, pericarditis, uveítis y miocarditis fatal [ver Reacciones adversas (Experiencia en estudios clínicos)].
Otras experiencias clínicas: A través de 21 ensayos de rango de dosis que administraron YERVOY® a dosis de 0,1 a 20 mg/kg (n=2478), también se reportaron las siguientes reacciones adversas mediadas por la respuesta inmunitaria probables con menos del 1% de incidencia: angiopatía, arteritis temporal, vasculitis, polimialgia reumática, conjuntivitis, blefaritis, epiescleritis, escleritis, iritis, vasculitis leucocitoclástica, eritema multiforme, psoriasis, artritis, tiroiditis autoinmune, hipoacusia neurosensorial, neuropatía autoinmune central (encefalitis), miositis, polimiositis, miositis ocular, anemia hemolítica y nefritis.
Toxicidad embriofetal: Basándose en su mecanismo de acción y los datos de estudios en animales, YERVOY® puede causar daño fetal cuando es administrado a una mujer embarazada. En los estudios de reproducción en animales, la administración de ipilimumab a macacos cynomolgus desde el inicio de la organogénesis hasta el parto dio lugar a una mayor incidencia de aborto, muerte fetal, parto prematuro (con un menor peso al nacer), y una mayor incidencia de la mortalidad infantil de una manera dosis-dependiente. Es probable que los efectos de ipilimumab sean mayores durante el segundo y tercer trimestre del embarazo. Asesorar a las mujeres embarazadas del riesgo potencial para el feto. Asesorar a las mujeres de potencial reproductivo de utilizar métodos anticonceptivos eficaces durante el tratamiento con un régimen que contenga YERVOY® y durante 3 meses después de la última dosis de YERVOY® [ver Uso en poblaciones específicas (Embarazo, Mujeres y hombres de potencial reproductivo)].
USO EN POBLACIONES ESPECÍFICAS:
Embarazo: Resumen de riesgos. Basado en datos de estudios en animales y su mecanismo de acción, YERVOY® puede causar daño fetal cuando se administra a una mujer embarazada [ver Farmacología clínica (Mecanismo de acción)]. En los estudios de reproducción en animales, la administración de ipilimumab a macacos desde el inicio de la organogénesis hasta el parto dio lugar a una mayor incidencia de aborto, muerte fetal, parto prematuro (con menor peso que el correspondiente al nacer), y una mayor incidencia de la mortalidad infantil en una manera dependiente de la dosis [ver Datos]. Es probable que los efectos de ipilimumab sean mayores durante el segundo y tercer trimestre del embarazo. La IgG1 humana es conocida por atravesar la barrera placentaria e ipilimumab es un IgG1; por lo tanto, ipilimumab tiene el potencial de ser transmitido de la madre al feto en desarrollo. Hay datos insuficientes para la exposición humana a YERVOY® en mujeres embarazadas. Asesorar a las mujeres embarazadas del riesgo potencial para el feto. En la población general de Estados Unidos, el riesgo de fondo estimado de los principales defectos de nacimiento y aborto involuntario en los embarazos clínicamente reconocidos son del 2% al 4% y del 15% a 20%, respectivamente.
Datos: Datos de animales. En un estudio combinado sobre el desarrollo embriofetal, perinatal y postnatal, se administró ipilimumab a ejemplares preñadas de Macacos cynomolgus cada 3 semanas a partir del inicio de la organogénesis en el primer trimestre hasta el parto. No se detectaron efectos adversos relacionados con el tratamiento sobre la reproducción durante los primeros dos trimestres del embarazo. A partir del tercer trimestre, la administración de ipilimumab a exposiciones de dosis de aproximadamente 2,6 a 7,2 veces de la exposición humana a una dosis de 3 mg/kg resultó en aumentos relacionados con la dosis en abortos, nacimientos de crías muertas, partos prematuros (con el correspondiente menor peso al nacimiento) y un incremento de incidencias de mortalidad infantil. Además, se identificaron anomalías del desarrollo en el sistema urogenital de 2 monos infantiles expuestos en el útero a 30 mg/kg de ipilimumab (7,2 veces el AUC en humanos a dosis de 3 mg/kg). Una mona hembra infante tuvo agenesia renal unilateral del riñón y uréter izquierdo, y 1 mono bebé de sexo masculino tuvo una uretra imperforada con obstrucción urinaria asociada y edema escrotal subcutáneo. Ratones genéticamente diseñados heterocigotos para CTLA-4 (CTLA-4 +/-), la diana de ipilimumab, parecieron sanos y dieron a luz a crías CTLA-4 +/- heterocigotos saludables. La unión de ratones heterocigotos CTLA-4 +/- también produjo descendencia deficiente en CTLA-4 (homocigoto negativo, CTLA-4 -/-). La descendencia homocigota negativa CTLA-4-/- se mostró sana al nacer, exhibió signos de enfermedad linfoproliferativa multiorgánica a las 2 semanas de edad, y todos murieron a las 3 a 4 semanas de edad con linfoproliferación masiva y destrucción de tejidos de múltiples órganos.
Lactancia: Resumen de riesgos: Se desconoce si YERVOY® se secreta a través de la leche humana. En monos, ipilimumab se presentó en la leche. No hay datos para evaluar los efectos de YERVOY® en la producción de la leche. Advertir a las mujeres de descontinuar la lactancia durante el tratamiento con YERVOY® y durante 3 meses después de la última dosis. Datos. En las monas hembras tratadas a niveles de dosis que dan como resultado exposiciones 2,6 y 7,2 veces mayores que aquellas alcanzadas en humanos con la dosis de 3 mg/kg, el ipilimumab estuvo presente en la leche en concentraciones de 0,1 mcg/mL y 0,4 mcg/mL, lo cual representa una proporción de hasta 0,3% de la concentración sérica de estado estable del fármaco.
Mujeres y hombres de potencial reproductivo:
Anticoncepción: Basándose en su mecanismo de acción, YERVOY® puede causar daño fetal cuando se administra a una mujer embarazada [ver Uso en poblaciones específicas (Embarazo)]. Asesorar a las mujeres en capacidad de reproducción a utilizar métodos anticonceptivos eficaces durante el tratamiento con YERVOY® y durante 3 meses después de la última dosis de YERVOY®.
Uso pediátrico: No se han establecido la seguridad ni la eficacia de YERVOY® en pacientes pediátricos.
Uso geriátrico: De los 511 pacientes que recibieron tratamiento con YERVOY® en el Estudio 1, el 28% tenían 65 años o más. No se observaron diferencias generales en la eficacia ni en la seguridad entre los pacientes de edad avanzada (65 años o más) y los pacientes más jóvenes (menores de 65 años). El Estudio 2 no incluyó un número suficiente de pacientes de 65 años o más para determinar si responden de manera diferente a los pacientes más jóvenes.
Insuficiencia renal: No se requiere ajuste de la dosis en pacientes con insuficiencia renal [ver Farmacología clínica (12.3)].
Insuficiencia hepática: No se requiere ajuste de la dosis en pacientes con insuficiencia hepática leve (bilirrubina total [TB] >1,0 a 1,5 veces el límite superior del rango normal [ULN] o AST >ULN). YERVOY® no ha sido estudiado en pacientes con insuficiencia hepática moderada (TB >1,5 a 3,0 veces ULN y cualquier valor de AST) o severa (TB >3 veces ULN y cualquier valor de AST). [Ver Farmacología clínica (12.3).]
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES:
YERVOY® (ipilimumab) Solución inyectable para infusión Intravenosa:
• En frasco ampolla/vial de 50 mg/10mL (5 mg/mL)
• En frasco ampolla/vial de 200 mg/40 mL (5 mg/mL)
SOBREDOSIS: No se dispone de información acerca de la sobredosis con YERVOY®.
DESCRIPCIÓN: YERVOY® (ipilimumab) es un anticuerpo monoclonal humano recombinante que se une al antígeno 4 asociado a linfocitos T citotóxicos (CTLA-4). El ipilimumab es una inmunoglobulina IgG1 K con un peso molecular aproximado de 148 kDa. El ipilimumab se produce en los cultivos celulares de los mamíferos (ovario de hámster chino). YERVOY® es una solución estéril, sin preservantes, transparente a ligeramente opalescente, incolora a amarilla pálida para infusión intravenosa, que puede contener una pequeña cantidad de partículas de ipilimumab amorfas visiblemente translúcidas a blancas. Se suministra en viales de un solo uso de 50 mg/10 mL y 200 mg/40 mL.
Cada mililitro contiene 5 mg de ipilimumab y los siguientes ingredientes inactivos: ácido dietilentriaminopentaacético (DTPA), manitol, polisorbato 80 (de origen vegetal), cloruro de sodio, tris clorhidrato, y agua para inyección, USP; hidróxido de sodio y ácido clorhídrico, cantidad suficiente para pH 7.0.
FARMACOLOGÍA CLÍNICA:
Mecanismo de acción: La CTLA-4 es un regulador negativo de la actividad de las células T. El ipilimumab es un anticuerpo monoclonal que se fija a la CTLA-4 y bloquea la interacción de la CTLA-4 con sus ligandos, CD80/CD86. Se ha demostrado que el bloqueo de la CTLA-4 incrementa la activación y proliferación de las células T, incluida la activación y la proliferación de células T efectoras infiltrantes del tumor. La inhibición de la señalización de CTLA-4 también puede reducir la función de las células T regulatorias, lo cual puede contribuir a un aumento general de la capacidad de respuesta de las células T, incluida la respuesta inmune antitumoral.
Farmacocinética: La farmacocinética (PK) del ipilimumab se estudió en 785 pacientes con melanoma no extirpable o metastásico que recibieron dosis de 0,3, 3 ó 10 mg/kg una vez cada 3 semanas con un total de 4 dosis. La PK del ipilimumab es lineal en el rango de dosis de 0,3 a 10 mg/kg. Luego de la administración de YERVOY® cada 3 semanas la acumulación sistémica era de 1,5 veces o menos. Las concentraciones en estado estacionario del ipilimumab se alcanzaron a la tercera dosis; la Cmín media en estado estacionario fue de 19,4 mcg/mL en 3 mg/kg y de 58,1 mcg/mL en 10 mg/kg cada 3 semanas. El valor medio (coeficiente porcentual de variación) basado en un análisis farmacocinético poblacional para la vida media terminal (t1/2) fue de 15,4 días (34%) y para el clearance fue de 16,8 mL/h (38%).
Poblaciones específicas: Los efectos de varias covariables de la farmacocinética de ipilimumab se evaluaron en análisis farmacocinéticos de la población. El clearance del ipilimumab aumentó junto con el incremento del peso corporal; sustentando la dosis recomendada basada en el peso corporal (mg/kg). Los siguientes factores no tuvieron efectos clínicamente importantes sobre el clearance del ipilimumab: edad (rango de 23 a 88 años), sexo, estado general, insuficiencia renal, insuficiencia hepática leve, terapia antineoplásica previa y niveles basales de lactato deshidrogenasa (LDH). El efecto de la raza no se examinó debido a una cantidad limitada de datos disponibles en grupos étnicos no caucásicos.
Insuficiencia renal: El efecto de la insuficiencia renal sobre el clearance del ipilimumab se evaluó en pacientes con insuficiencia renal leve (GFR <90 y ≥60 mL/min/1,73 m2; n=349), moderada (GFR <60 y ≥30 mL/min/1,73 m2; n=82) o severa (GFR <30 y ≥15 mL/min/1,73 m2; n=4) en comparación con pacientes con función renal normal (GFR ≥90 mL/min/1,73 m2; n=350) en análisis farmacocinéticos poblacionales. No se hallaron diferencias clínicamente importantes en el clearance del ipilimumab entre pacientes con insuficiencia renal y pacientes con una función renal normal [ver uso en Poblaciones específicas (Insuficiencia hepática)].
Insuficiencia hepática: El efecto de la insuficiencia hepática sobre el clearance del ipilimumab se evaluó en pacientes con insuficiencia hepática leve (n=76) en comparación con pacientes con función hepática normal (n=708) en los análisis farmacocinéticos poblacionales y no se hallaron diferencias clínicamente importantes en el clearance del ipilimumab. YERVOY® no ha sido estudiado en pacientes con insuficiencia hepática moderada o severa [ver Uso en poblaciones específicas (8.7)].
INFORMACIÓN DE ASESORAMIENTO PARA EL PACIENTE:
Indicar al paciente que lea el folleto para el paciente aprobado por la FDA (Guía del Medicamento).
Reacciones adversas mediadas por la respuesta inmunitaria: Informar a los pacientes acerca del riesgo potencial de reacciones adversas mediadas por la respuesta inmunitaria [ver Advertencias y precauciones (Enterocolitis mediada por la respuesta inmunitaria, Hepatitis mediada por la respuesta inmunitaria, Dermatitis mediada por la respuesta inmunitaria, Neuropatías mediadas por la respuesta inmunitaria, Endocrinopatías mediadas por la respuesta inmunitaria, Otras reacciones adversas mediadas por la respuesta inmunitaria, incluidas las manifestaciones oculares)].
Toxicidad embriofetal: Asesorar a las pacientes mujeres que YERVOY® puede causar daño fetal. Asesorar a las mujeres de potencial reproductivo a utilizar métodos anticonceptivos eficaces durante el tratamiento con YERVOY® y durante 3 meses después de la última dosis. Asesorar a pacientes de sexo femenino que contacten con su médico en caso de que haya un embarazo conocido o sospechado.
Lactancia: Advertir a las mujeres en período de lactancia que no deben amamantar a sus hijos durante el tratamiento con YERVOY® y por 3 meses después de la última dosis [ver Uso en poblaciones específicas (Lactancia. Resumen de riesgos)].
GUÍA DE MEDICACIÓN PARA EL PACIENTE:
Lea esta Guía de Medicación para el Paciente antes de comenzar a recibir YERVOY® y antes de cada infusión. Puede contener información nueva. Esta Guía de Medicación para el Paciente no reemplaza la consulta con su médico respecto de su afección médica o su tratamiento.
¿Cuál es la información más importante que debo conocer acerca de YERVOY®?: YERVOY® puede causar efectos secundarios graves en muchas partes de su cuerpo que pueden producir la muerte. Estos problemas pueden aparecer en cualquier momento durante el tratamiento con YERVOY® o después que haya concluido el tratamiento. Llame a su médico de inmediato si presenta cualquiera de estos signos o síntomas, o empeoran. No intente tratar los síntomas usted mismo. Inflamación intestinal (colitis) que puede causar desgarros o agujeros (perforación) en los intestinos. Los signos y síntomas de la colitis pueden incluir:
• Diarrea (heces blandas) o más movimientos intestinales de lo normal.
• Sangre en las heces o heces oscuras, alquitranadas, pegajosas.
• Dolor estomacal (dolor abdominal) o dolor a la palpación.
Problemas hepáticos (hepatitis) que pueden producir insuficiencia hepática: Los signos y síntomas de la hepatitis pueden incluir:
• Color amarillo en la piel o en la parte blanca de los ojos
• Orina oscura (color té)
• Náuseas o vómitos
• Dolor en el lado derecho del estómago
• Hemorragias o hematomas que se producen con más facilidad de lo normal
Problemas cutáneos que pueden causar una reacción cutánea grave: Los signos y síntomas de las reacciones cutáneas graves pueden incluir:
• Erupción cutánea con picazón o sin picazón
• Llagas en la boca
• Ampollas y/o peladuras en la piel
Problemas nerviosos que pueden provocar parálisis: Los síntomas de los problemas de los nervios pueden incluir:
• Debilidad inusual de las piernas, los brazos o la cara
• Entumecimiento u hormigueo de las manos o los pies
Problemas de las glándulas hormonales (en especial la pituitaria, la suprarrenal y la tiroides) que pueden afectar su funcionamiento: Los signos y síntomas que indican que las glándulas no están funcionando bien pueden incluir los siguientes:
• Dolor de cabeza persistente o inusual
• Pereza inusual
• Sensación de frío permanente
• Aumento de peso
• Cambios de humor o del comportamiento, como deseo sexual disminuido, irritabilidad u olvidos
• Mareos o desmayos
Problemas oculares: Los síntomas pueden incluir:
• Visión borrosa, visión doble u otros problemas de la visión
• Dolor o enrojecimiento ocular
Obtener tratamiento médico de inmediato puede evitar que el problema se agrave: Su médico le examinará para estos problemas durante el tratamiento con YERVOY®. Su médico le puede tratar con medicamentos corticosteroides. Su médico puede tener que retrasar o interrumpir el tratamiento por completo con YERVOY® si usted tiene efectos secundarios graves.
¿Qué es YERVOY®?
YERVOY® es un medicamento de venta bajo receta que se utiliza en adultos para el tratamiento del melanoma (un tipo de cáncer de la piel). YERVOY® puede ser usado:
• Cuando el melanoma se ha extendido y no puede extirparse quirúrgicamente.
• Para prevenir que el melanoma regrese luego de que los ganglios linfáticos que contienen células cancerosas hayan sido removidos mediante cirugía.
No se sabe si YERVOY® es seguro y efectivo en niños menores de 18 años de edad.
¿Qué debo decirle a mi médico antes de recibir YERVOY®?
Antes de recibir YERVOY®, debe informar a su médico acerca de sus problemas de salud, incluyendo si usted:
• Padece una afección del sistema inmunitario (enfermedad autoinmunitaria), como colitis ulcerosa, enfermedad de Crohn, lupus, o sarcoidosis
• Recibió un trasplante de órgano
• Padece daño hepático
• Está embarazada o tiene planes de quedar embarazada. YERVOY® puede dañar a su bebé antes de su nacimiento.
– Las mujeres que son capaces de quedar embarazadas deben utilizar métodos anticonceptivos eficaces durante el tratamiento con YERVOY® y durante 3 meses después de la última dosis de YERVOY®.
• Está dando de lactar o planea dar de lactar.
– No se sabe si YERVOY® se pasa mediante la leche materna.
– No amamantar durante el tratamiento con YERVOY® y por 3 meses después de la última dosis de YERVOY®.
Informe a su médico acerca de todos los medicamentos que toma, incluidos todos los medicamentos de venta bajo receta y de venta libre, vitaminas, y suplementos a base de hierbas. Usted debe conocer los medicamentos que toma. Confeccione una lista para mostrársela a sus médicos y farmacéuticos cada vez que compra un medicamento nuevo. No debe comenzar un tratamiento con un medicamento nuevo antes de hablar con el médico que le recetó YERVOY®.
¿Cómo se me administrarán las dosis de YERVOY®?
• Se le administrará YERVOY® a través de una vía intravenosa (IV) en su vena por 90 minutos.
• Su médico decidirá cuántos tratamientos usted va a necesitar.
• Su médico le realizará exámenes de sangre antes y durante el tratamiento con YERVOY®.
• Es importante que usted mantenga todas las citas con su médico. Llame a su médico si usted olvida una cita. Puede haber instrucciones especiales para usted.
¿Cuáles son los efectos secundarios que puede provocar YERVOY®?
YERVOY® puede provocar efectos secundarios graves. Lea “¿Cuál es la información más importante que debo conocer acerca de YERVOY®?”
Los efectos secundarios más comunes de YERVOY® incluyen:
• Cansancio
• Diarrea
• Picazón
• Erupción cutánea
• Náuseas
• Vómitos
• Dolor de cabeza
• Pérdida de peso
• Fiebre
• Disminución del apetito
• Dificultad para dormir o permanecer dormido
Dígale a su médico si usted tiene algún efecto secundario que le molesta o que no desaparece. Estos no son todos los efectos secundarios posibles de YERVOY®. Para obtener más información, consulte a su médico. Comuníquese con su médico con el fin de recibir asesoramiento médico acerca de los efectos secundarios.
Información general acerca del uso seguro y eficaz de YERVOY®:
En ocasiones, los medicamentos se recetan con fines que no están enumerados en la Guía de Medicación para el Paciente. Esta Guía de Medicación para el Paciente resume la información más importante acerca de YERVOY®. Si desea obtener más información, consulte a su médico. Puede pedirle a su médico que le proporcione información acerca de YERVOY® orientada específicamente a los médicos.
¿Cuáles son los ingredientes de YERVOY®?
Principio activo: Ipilimumab; excipientes c.s.
Consulte a su médico.
Fabricado por: Baxter Pharmaceutical Solutions, LLC
Indiana, EEUU
Para: Bristol-Myers Squibb Company – EEUU
Importado por:
BRISTOL-MYERS SQUIBB PERÚ, S.A.
Av. Canaval y Moreyra 380, San Isidro
Lima, Perú


